

Genetic characterization of norovirus GII.4 variants circulating in Canada using a metagenomic technique
- PMID: 30333011
- PMCID: PMC6191920
- DOI: 10.1186/s12879-018-3419-8
Genetic characterization of norovirus GII.4 variants circulating in Canada using a metagenomic technique
Abstract
Background: Human norovirus is the leading cause of viral gastroenteritis globally, and the GII.4 has been the most predominant genotype for decades. This genotype has numerous variants that have caused repeated epidemics worldwide. However, the molecular evolutionary signatures among the GII.4 variants have not been elucidated throughout the viral genome.
Method: A metagenomic, next-generation sequencing method, based on Illumina RNA-Seq, was applied to determine norovirus sequences from clinical samples.
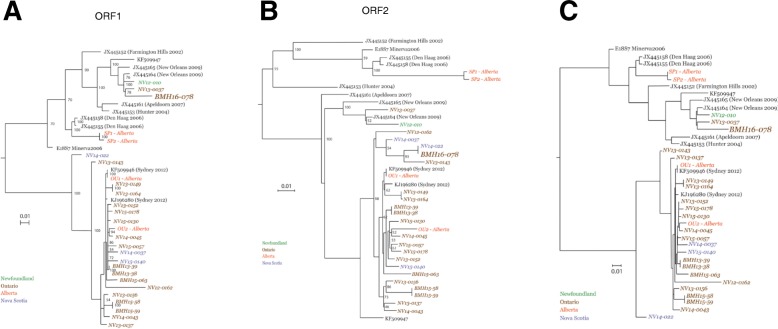
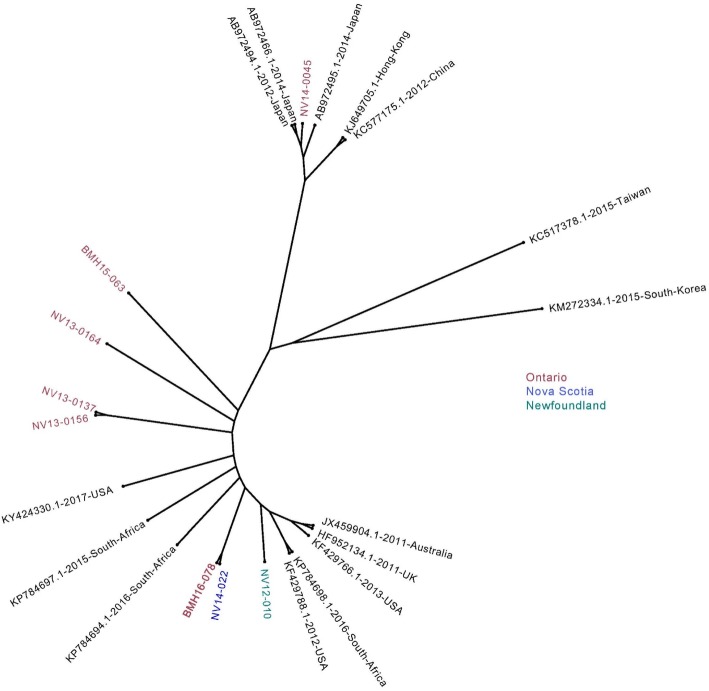
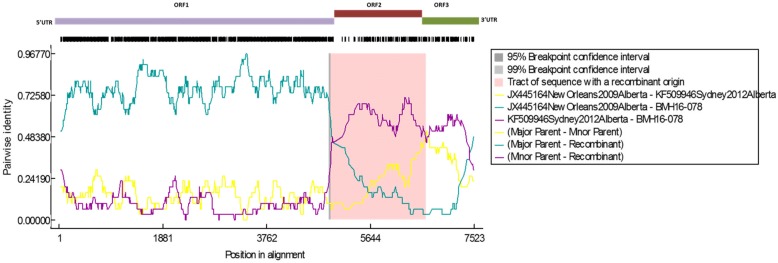
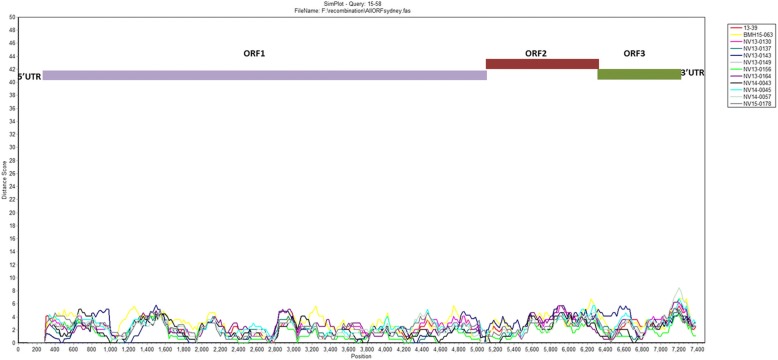
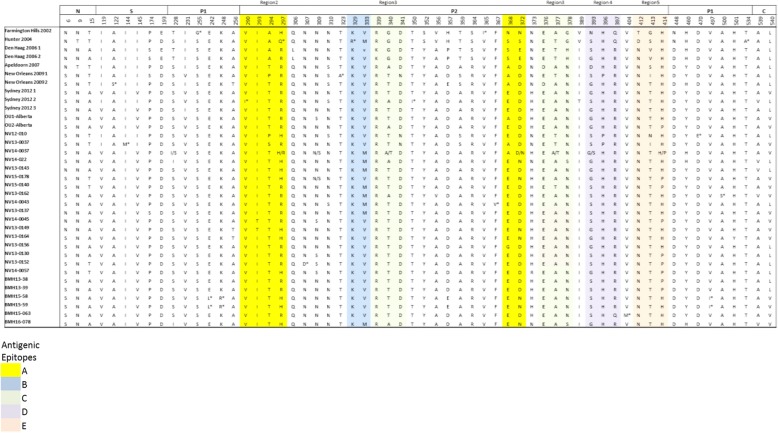
Results: Herein, the obtained deep-sequencing data was employed to analyze full-genomic sequences from GII.4 variants prevailing in Canada from 2012 to 2016. Phylogenetic analysis demonstrated that the majority of these sequences belong to New Orleans 2009 and Sydney 2012 strains, and a recombinant sequence was also identified. Genome-wide similarity analyses implied that while the capsid gene is highly diverse among the isolates, the viral protease and polymerase genes remain relatively conserved. Numerous amino acid substitutions were observed at each putative antigenic epitope of the VP1 protein, whereas few amino acid changes were identified in the polymerase protein. Co-infection with other enteric RNA viruses was investigated and the astrovirus genome was identified in one of the samples.
Conclusions: Overall this study demonstrated the application of whole genome sequencing as an important tool in molecular characterization of noroviruses.
Keywords: Antigenic drift; Co-infection; Metagenomics; Next-generation sequencing; Norovirus; Recombination.
Conflict of interest statement
Ethics approval and consent to participate
This study has been granted an exemption from requiring ethics approval by Health Canada and a formal consent was not required because the study participants were anonymized.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Havelaar Arie H., Kirk Martyn D., Torgerson Paul R., Gibb Herman J., Hald Tine, Lake Robin J., Praet Nicolas, Bellinger David C., de Silva Nilanthi R., Gargouri Neyla, Speybroeck Niko, Cawthorne Amy, Mathers Colin, Stein Claudia, Angulo Frederick J., Devleesschauwer Brecht. World Health Organization Global Estimates and Regional Comparisons of the Burden of Foodborne Disease in 2010. PLOS Medicine. 2015;12(12):e1001923. doi: 10.1371/journal.pmed.1001923. - DOI - PMC - PubMed
-
- de Graaf M, van Beek J, Koopmans MP. Human norovirus transmission and evolution in a changing world. Nat Rev Microbiol. 2016. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
