

Chromosome evolution at the origin of the ancestral vertebrate genome
- PMID: 30333059
- PMCID: PMC6193309
- DOI: 10.1186/s13059-018-1559-1
Chromosome evolution at the origin of the ancestral vertebrate genome
Abstract
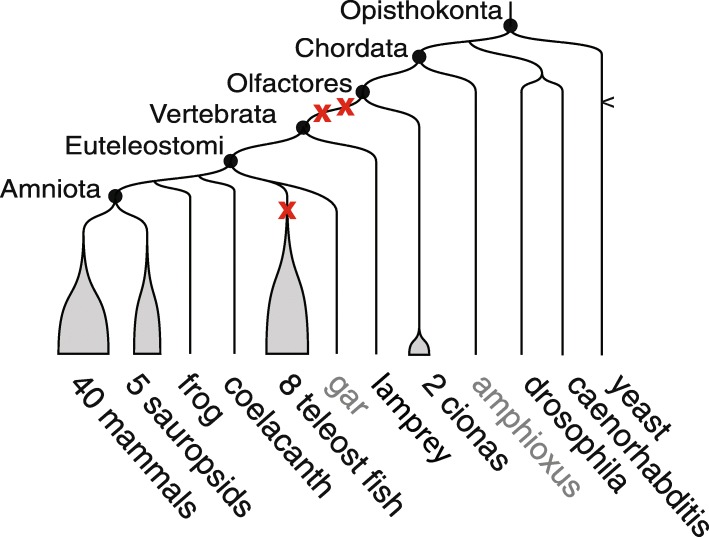
Background: It has been proposed that more than 450 million years ago, two successive whole genome duplications took place in a marine chordate lineage before leading to the common ancestor of vertebrates. A precise reconstruction of these founding events would provide a framework to better understand the impact of these early whole genome duplications on extant vertebrates.
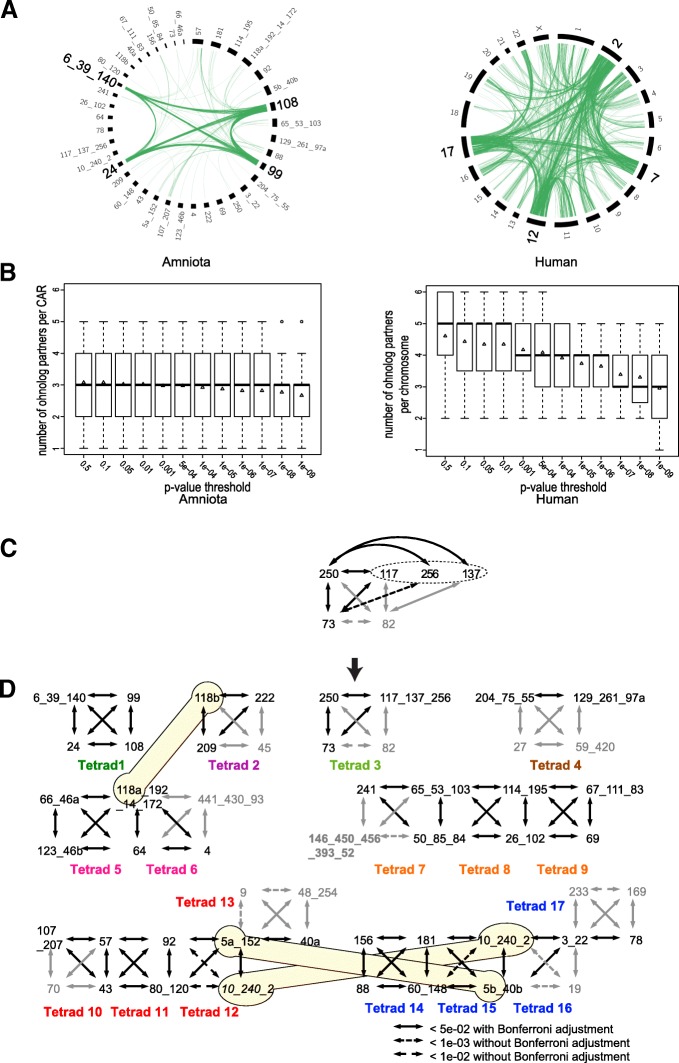
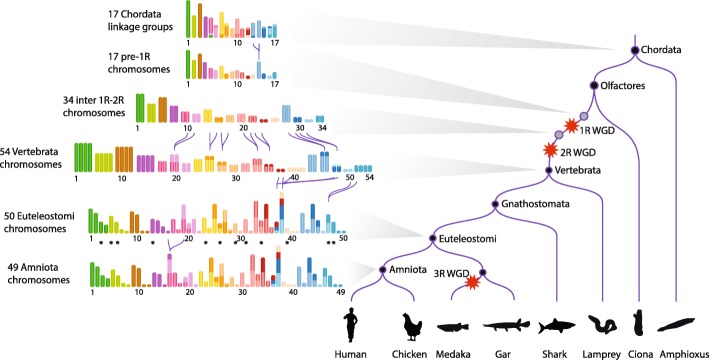
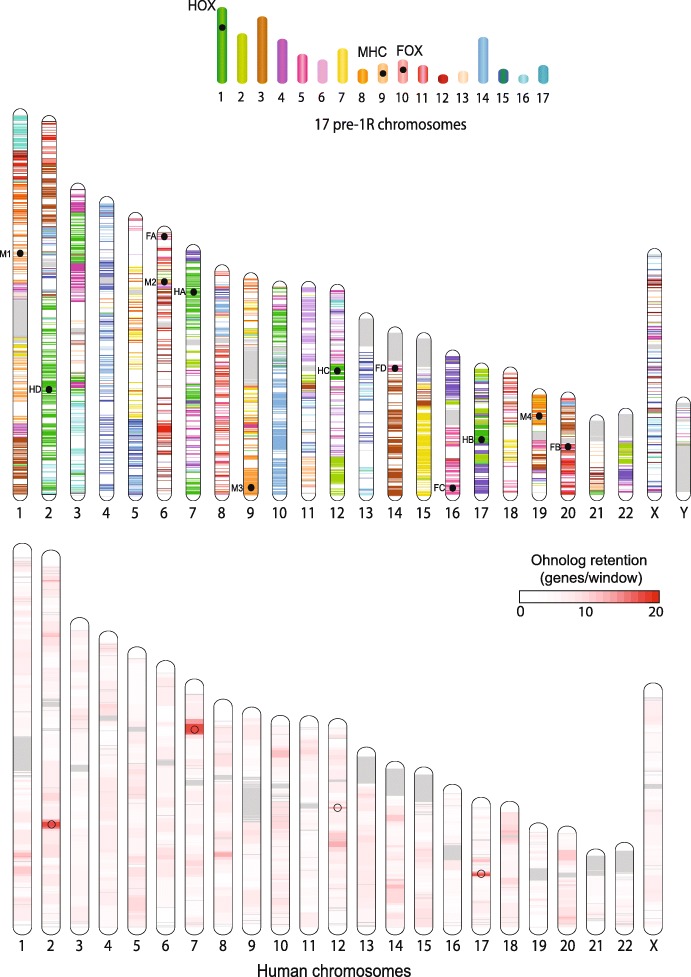
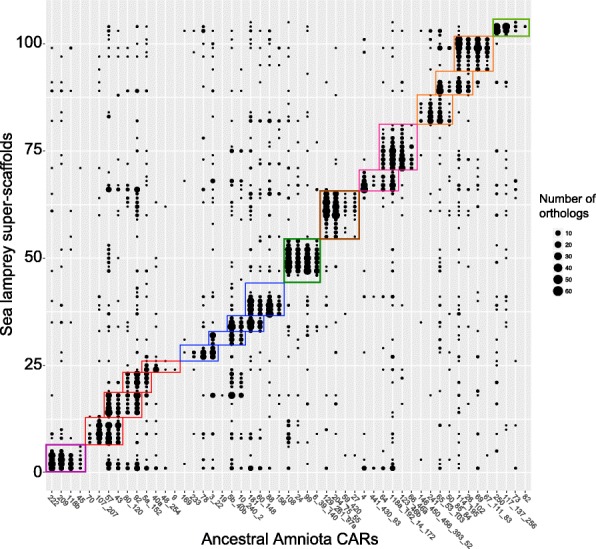
Results: We reconstruct the evolution of chromosomes at the beginning of vertebrate evolution. We first compare 61 extant animal genomes to reconstruct the highly contiguous order of genes in a 326-million-year-old ancestral Amniota genome. In this genome, we establish a well-supported list of duplicated genes originating from the two whole genome duplications to identify tetrads of duplicated chromosomes. From this, we reconstruct a chronology in which a pre-vertebrate genome composed of 17 chromosomes duplicated to 34 chromosomes and was subject to seven chromosome fusions before duplicating again into 54 chromosomes. After the separation of the lineage of Gnathostomata (jawed vertebrates) from Cyclostomata (extant jawless fish), four more fusions took place to form the ancestral Euteleostomi (bony vertebrates) genome of 50 chromosomes.
Conclusions: These results firmly establish the occurrence of two whole genome duplications in the lineage that precedes the ancestor of vertebrates, resolving in particular the ambiguity raised by the analysis of the lamprey genome. This work provides a foundation for studying the evolution of vertebrate chromosomes from the standpoint of a common ancestor and particularly the pattern of duplicate gene retention and loss that resulted in the gene composition of extant vertebrate genomes.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures


Comment in
-
A new look at an old question: when did the second whole genome duplication occur in vertebrate evolution?Genome Biol. 2018 Nov 28;19(1):209. doi: 10.1186/s13059-018-1592-0. Genome Biol. 2018. PMID: 30486862 Free PMC article.
References
-
- Ohno S. Evolution by gene duplication. London: Allen and Unwin; 1970.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
