

Stress response protein GJA1-20k promotes mitochondrial biogenesis, metabolic quiescence, and cardioprotection against ischemia/reperfusion injury
- PMID: 30333316
- PMCID: PMC6237442
- DOI: 10.1172/jci.insight.121900
Stress response protein GJA1-20k promotes mitochondrial biogenesis, metabolic quiescence, and cardioprotection against ischemia/reperfusion injury
Abstract
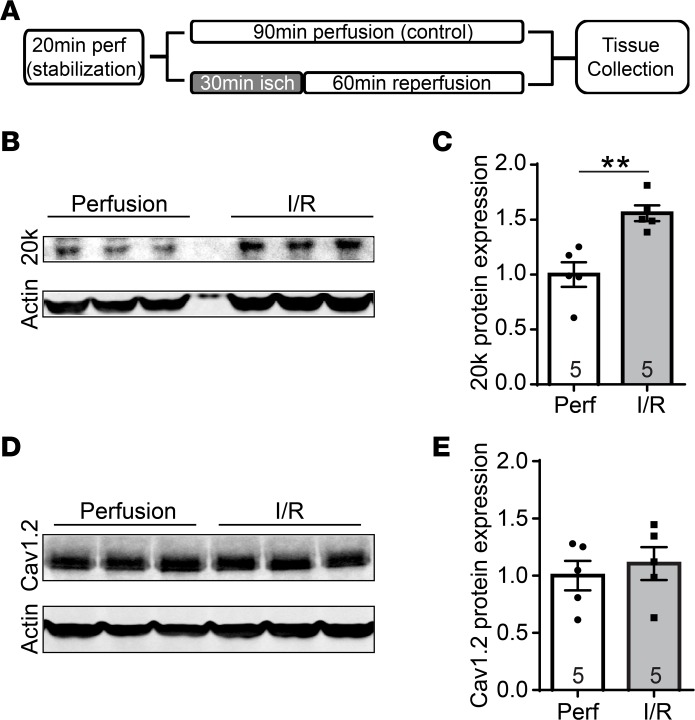
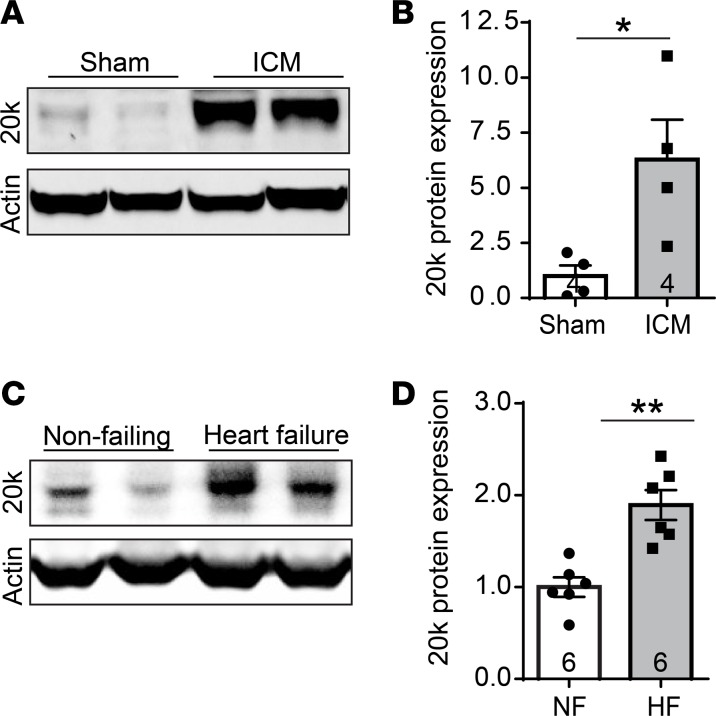
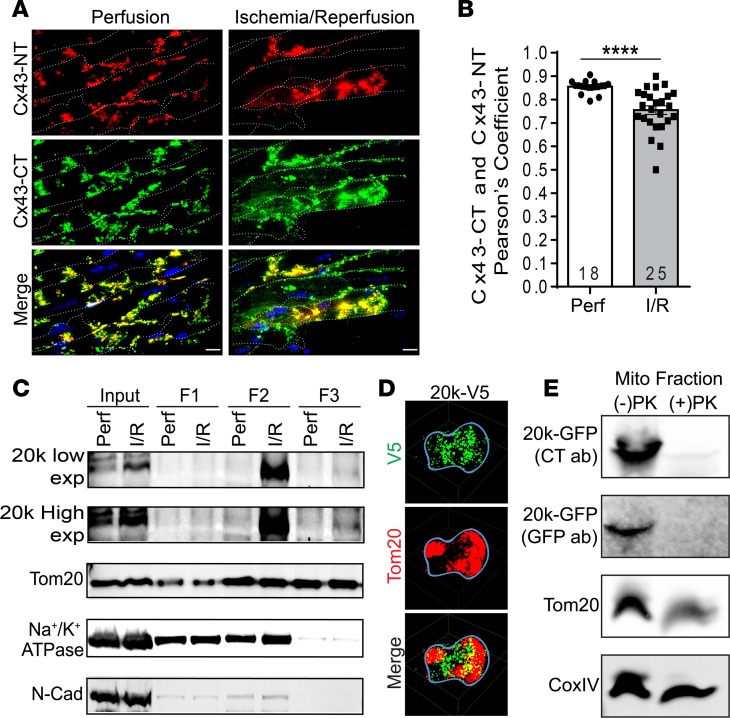
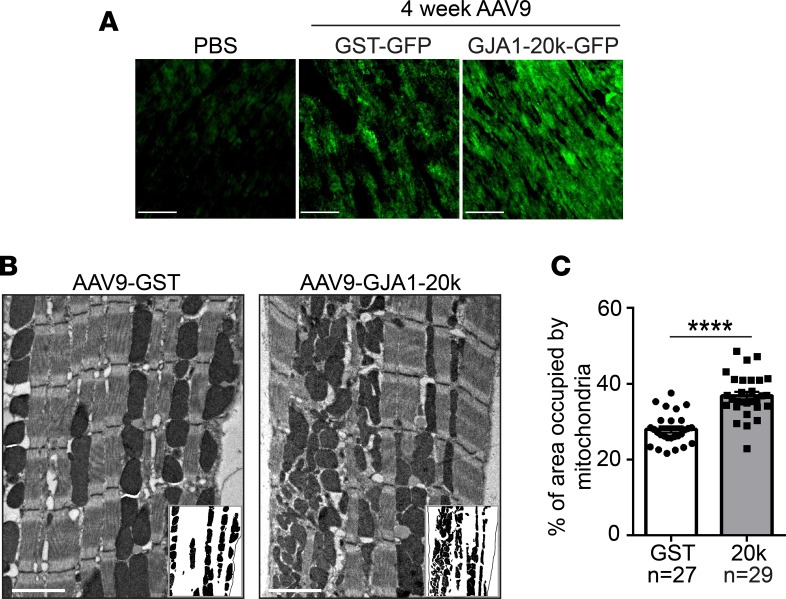
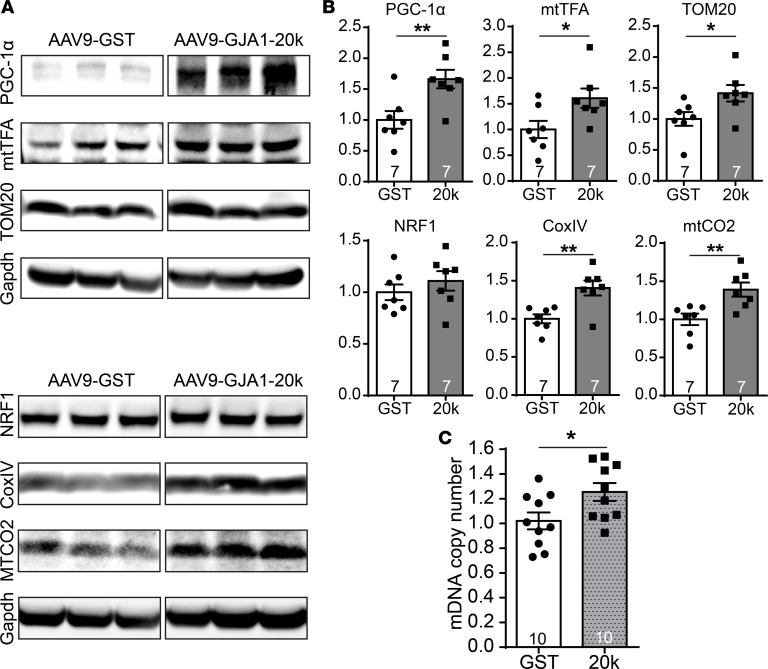
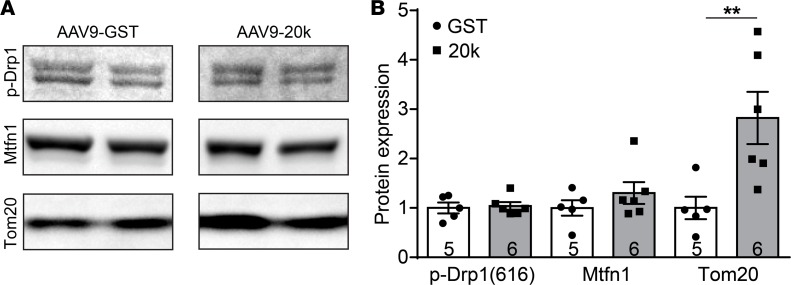
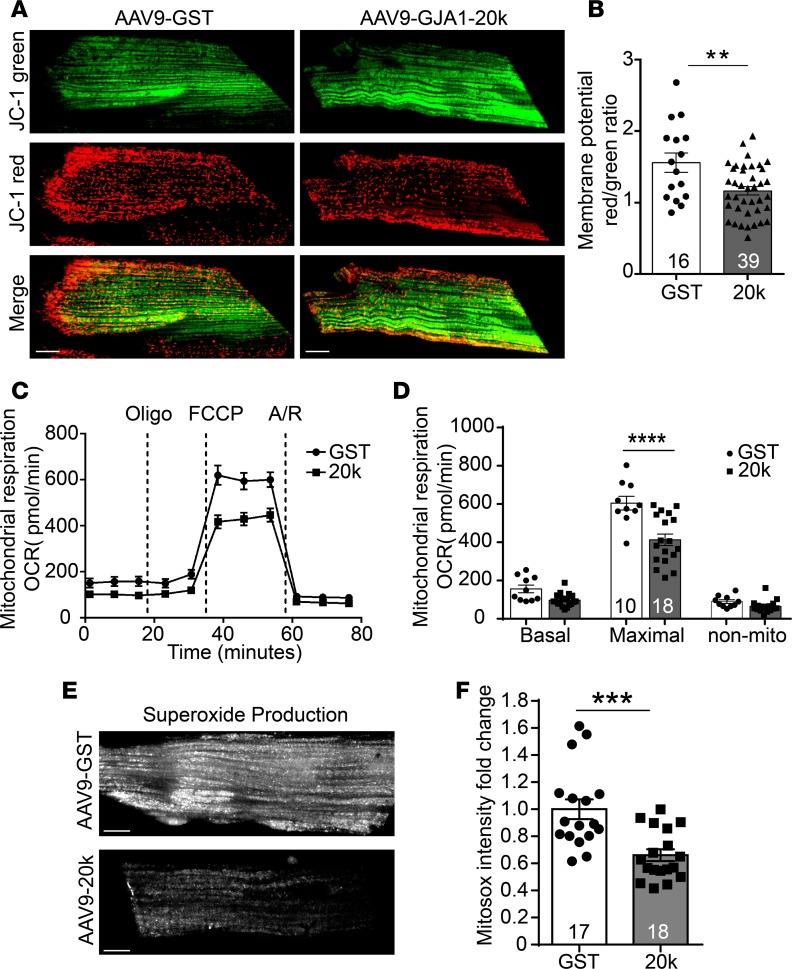
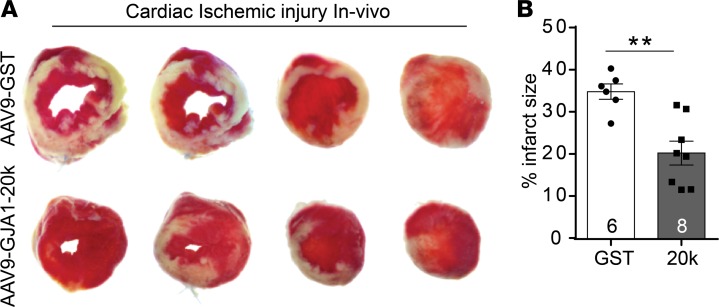
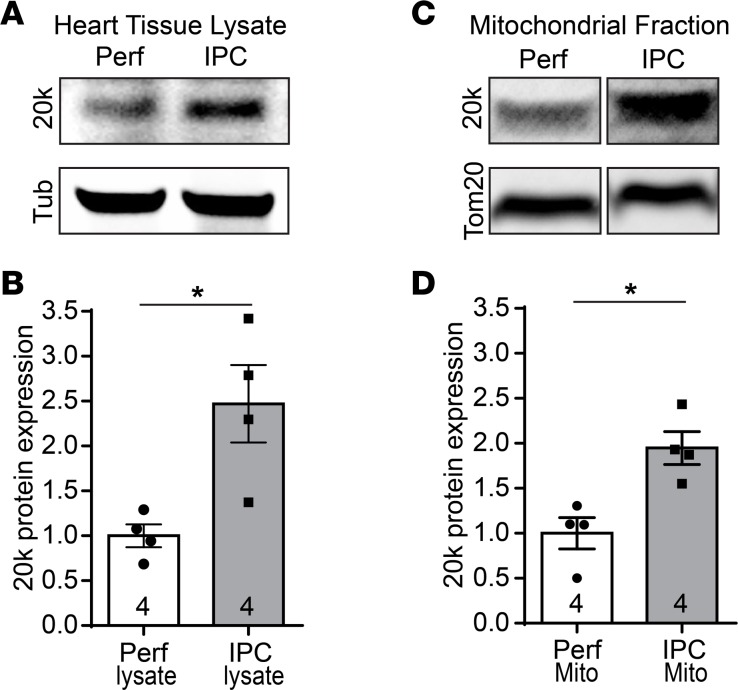
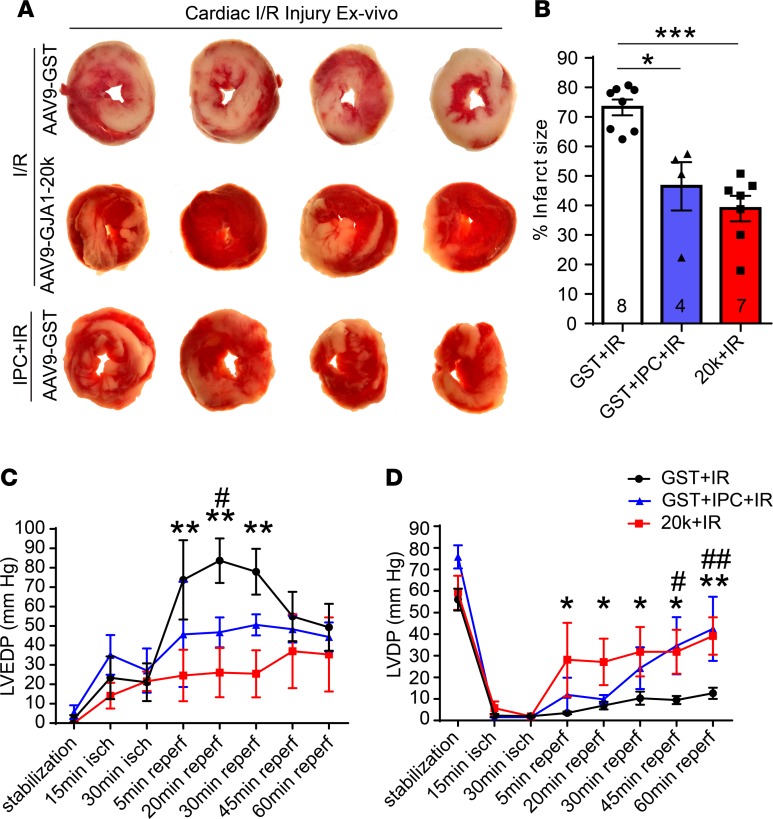
Connexin 43 (Cx43), a product of the GJA1 gene, is a gap junction protein facilitating intercellular communication between cardiomyocytes. Cx43 protects the heart from ischemic injury by mechanisms that are not well understood. GJA1 mRNA can undergo alternative translation, generating smaller isoforms in the heart, with GJA1-20k being the most abundant. Here, we report that ischemic and ischemia/reperfusion (I/R) injuries upregulate endogenous GJA1-20k protein in the heart, which targets to cardiac mitochondria and associates with the outer mitochondrial membrane. Exploring the functional consequence of increased GJA1-20k, we found that AAV9-mediated gene transfer of GJA1-20k in mouse hearts increases mitochondrial biogenesis while reducing mitochondrial membrane potential, respiration, and ROS production. By doing so, GJA1-20k promotes a protective mitochondrial phenotype, as seen with ischemic preconditioning (IPC), which also increases endogenous GJA1-20k in heart lysates and mitochondrial fractions. As a result, AAV9-GJA1-20k pretreatment reduces myocardial infarct size in mouse hearts subjected to in vivo ischemic injury or ex vivo I/R injury, similar to an IPC-induced cardioprotective effect. In conclusion, GJA1-20k is an endogenous stress response protein that induces mitochondrial biogenesis and metabolic hibernation, preconditioning the heart against I/R insults. Introduction of exogenous GJA1-20k is a putative therapeutic strategy for patients undergoing anticipated ischemic injury.
Keywords: Heart failure; Ion channels; Metabolism; Muscle Biology; Translation.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
