

NTRK fusion-positive cancers and TRK inhibitor therapy
- PMID: 30333516
- PMCID: PMC6419506
- DOI: 10.1038/s41571-018-0113-0
NTRK fusion-positive cancers and TRK inhibitor therapy
Abstract
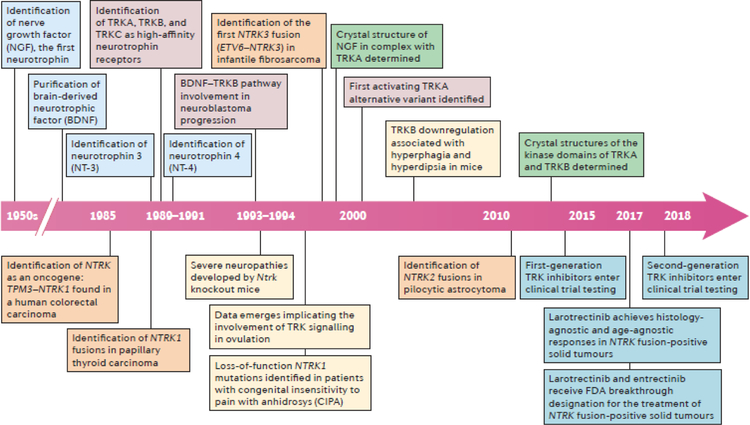
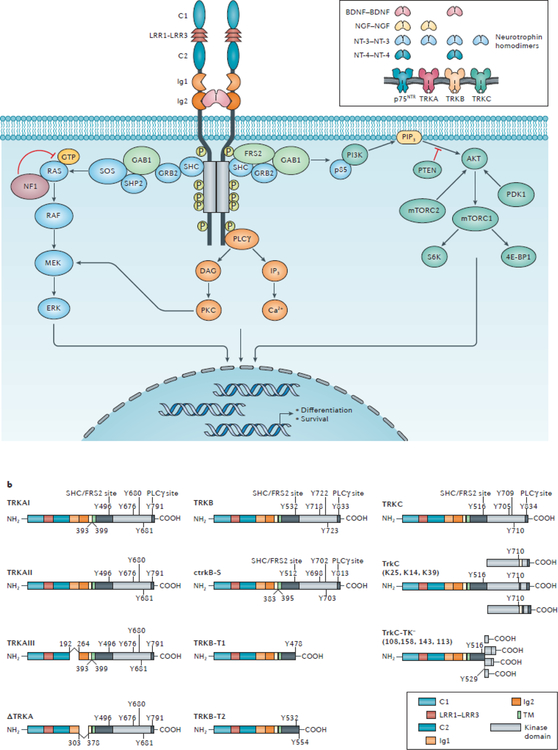
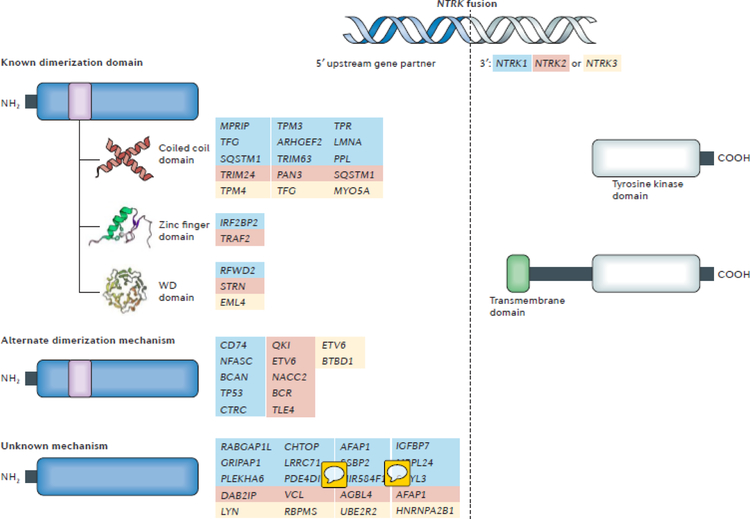
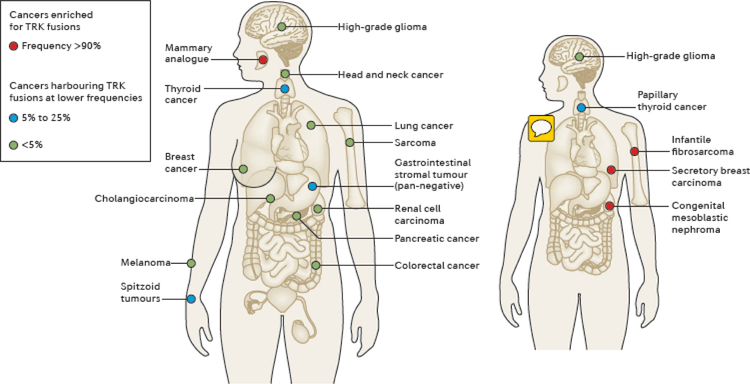
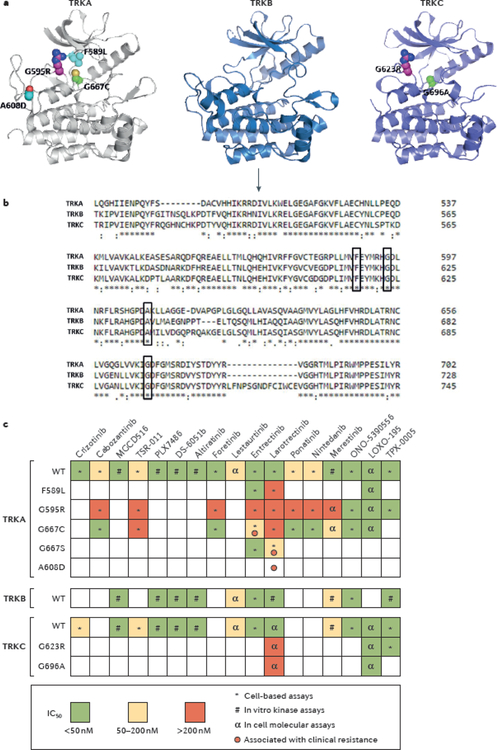
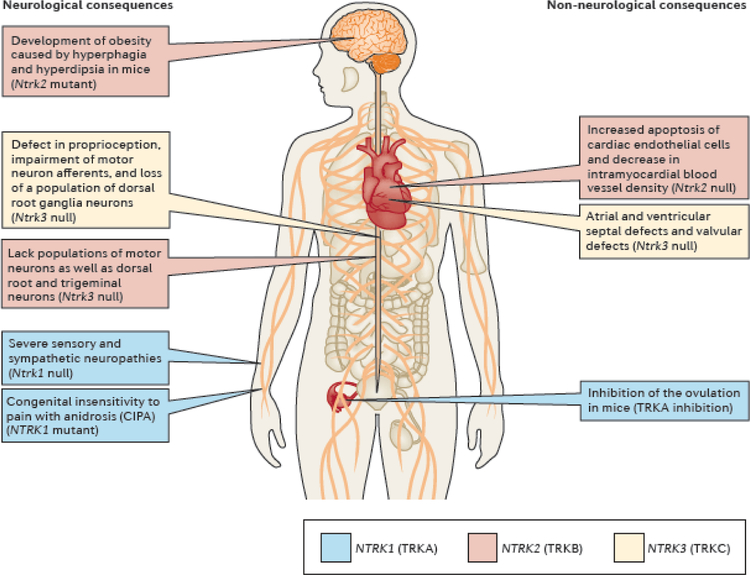
NTRK gene fusions involving either NTRK1, NTRK2 or NTRK3 (encoding the neurotrophin receptors TRKA, TRKB and TRKC, respectively) are oncogenic drivers of various adult and paediatric tumour types. These fusions can be detected in the clinic using a variety of methods, including tumour DNA and RNA sequencing and plasma cell-free DNA profiling. The treatment of patients with NTRK fusion-positive cancers with a first-generation TRK inhibitor, such as larotrectinib or entrectinib, is associated with high response rates (>75%), regardless of tumour histology. First-generation TRK inhibitors are well tolerated by most patients, with toxicity profiles characterized by occasional off-tumour, on-target adverse events (attributable to TRK inhibition in non-malignant tissues). Despite durable disease control in many patients, advanced-stage NTRK fusion-positive cancers eventually become refractory to TRK inhibition; resistance can be mediated by the acquisition of NTRK kinase domain mutations. Fortunately, certain resistance mutations can be overcome by second-generation TRK inhibitors, including LOXO-195 and TPX-0005 that are being explored in clinical trials. In this Review, we discuss the biology of NTRK fusions, strategies to target these drivers in the treatment-naive and acquired-resistance disease settings, and the unique safety profile of TRK inhibitors.
Conflict of interest statement
Competing interests
A.D. has received honoraria (as an advisory board member) from Bayer, Ignyta, Loxo Oncology, Pfizer, Roche/Genentech, and TP Therapeutics, and research funding from Loxo Oncology. M.S. has received research funding from Daiichi Sankyo and Puma Biotechnology.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
