

Modeling Congenital Disorders of N-Linked Glycoprotein Glycosylation in Drosophila melanogaster
- PMID: 30333856
- PMCID: PMC6176275
- DOI: 10.3389/fgene.2018.00436
Modeling Congenital Disorders of N-Linked Glycoprotein Glycosylation in Drosophila melanogaster
Abstract
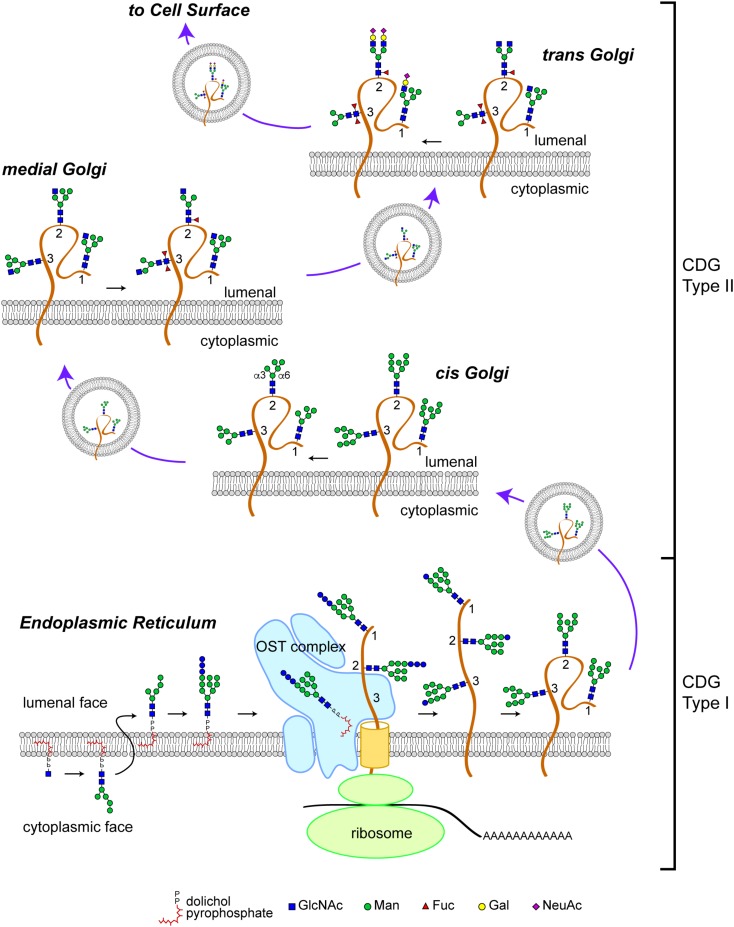
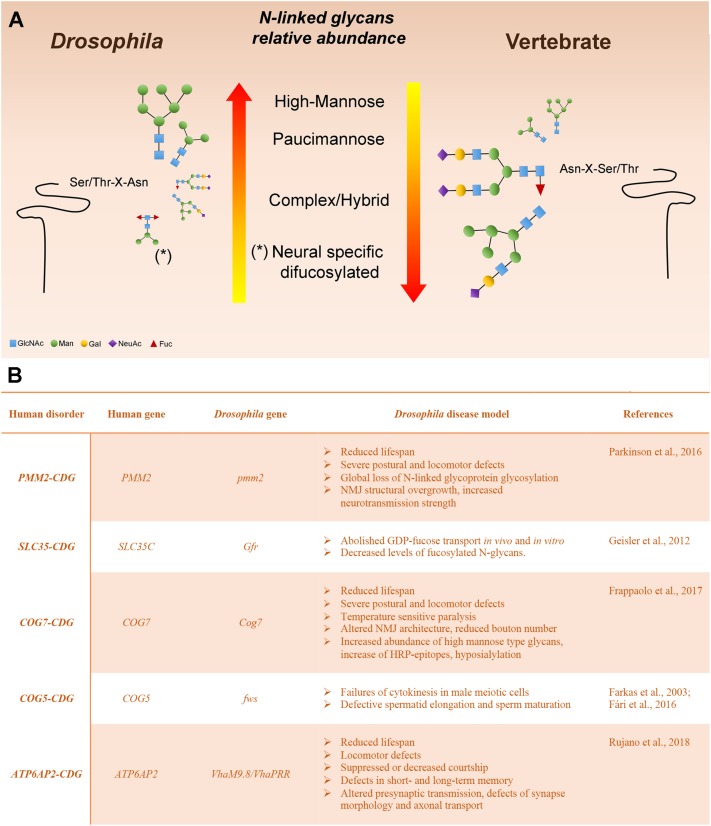
Protein glycosylation, the enzymatic addition of N-linked or O-linked glycans to proteins, serves crucial functions in animal cells and requires the action of glycosyltransferases, glycosidases and nucleotide-sugar transporters, localized in the endoplasmic reticulum and Golgi apparatus. Congenital Disorders of Glycosylation (CDGs) comprise a family of multisystemic diseases caused by mutations in genes encoding proteins involved in glycosylation pathways. CDGs are classified into two large groups. Type I CDGs affect the synthesis of the dolichol-linked Glc3Man9GlcNac2 precursor of N-linked glycosylation or its transfer to acceptor proteins. Type II CDG (CDG-II) diseases impair either the trimming of the N-linked oligosaccharide, the addition of terminal glycans or the biosynthesis of O-linked oligosaccharides, which occur in the Golgi apparatus. So far, over 100 distinct forms of CDGs are known, with the majority of them characterized by neurological defects including mental retardation, seizures and hypotonia. Yet, it is unclear how defective glycosylation causes the pathology of CDGs. This issue can be only addressed by developing animal models of specific CDGs. Drosophila melanogaster is emerging as a highly suitable organism for analyzing glycan-dependent functions in the central nervous system (CNS) and the involvement of N-glycosylation in neuropathologies. In this review we illustrate recent work that highlights the genetic and neurobiologic advantages offered by D. melanogaster for dissecting glycosylation pathways and modeling CDG pathophysiology.
Keywords: Drosophila; Golgi; congenital disorders; glycosylation; model organism.
Figures
Similar articles
-
Komrower Lecture. Congenital disorders of glycosylation (CDG): it's all in it!J Inherit Metab Dis. 2003;26(2-3):99-118. doi: 10.1023/a:1024431131208. J Inherit Metab Dis. 2003. PMID: 12889654
-
Abnormal synthesis of dolichol-linked oligosaccharides in carbohydrate-deficient glycoprotein syndrome.Glycobiology. 1995 Jul;5(5):503-10. doi: 10.1093/glycob/5.5.503. Glycobiology. 1995. PMID: 8563136
-
[Congenital disorders of glycosylation: state of the art and Spanish experience].Med Clin (Barc). 2004 May 15;122(18):707-16. doi: 10.1016/s0025-7753(04)74362-6. Med Clin (Barc). 2004. PMID: 15171833 Review. Spanish.
-
Modeling human congenital disorder of glycosylation type IIa in the mouse: conservation of asparagine-linked glycan-dependent functions in mammalian physiology and insights into disease pathogenesis.Glycobiology. 2001 Dec;11(12):1051-70. doi: 10.1093/glycob/11.12.1051. Glycobiology. 2001. PMID: 11805078
-
[Congenital disorders of glycosylation].Ann Pharm Fr. 2003;61(5):330-9. Ann Pharm Fr. 2003. PMID: 13130291 Review. French.
Cited by
-
Serum N-Glycome Diversity in Teleost and Chondrostrean Fishes.Front Mol Biosci. 2021 Nov 10;8:778383. doi: 10.3389/fmolb.2021.778383. eCollection 2021. Front Mol Biosci. 2021. PMID: 34859056 Free PMC article.
-
The Close Relationship between the Golgi Trafficking Machinery and Protein Glycosylation.Cells. 2020 Dec 10;9(12):2652. doi: 10.3390/cells9122652. Cells. 2020. PMID: 33321764 Free PMC article. Review.
-
CNS glycosylphosphatidylinositol deficiency results in delayed white matter development, ataxia and premature death in a novel mouse model.Hum Mol Genet. 2020 May 8;29(7):1205-1217. doi: 10.1093/hmg/ddaa046. Hum Mol Genet. 2020. PMID: 32179897 Free PMC article.
-
Case report: The art of anesthesiology-Approaching a minor procedure in a child with MPI-CDG.Front Pharmacol. 2022 Dec 15;13:1038090. doi: 10.3389/fphar.2022.1038090. eCollection 2022. Front Pharmacol. 2022. PMID: 36588700 Free PMC article.
-
Microtubule and Actin Cytoskeletal Dynamics in Male Meiotic Cells of Drosophila melanogaster.Cells. 2022 Feb 16;11(4):695. doi: 10.3390/cells11040695. Cells. 2022. PMID: 35203341 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources
Molecular Biology Databases