

BH3-Dependent and Independent Activation of BAX and BAK in Mitochondrial Apoptosis
- PMID: 30334018
- PMCID: PMC6186458
- DOI: 10.1016/j.cophys.2018.03.005
BH3-Dependent and Independent Activation of BAX and BAK in Mitochondrial Apoptosis
Abstract
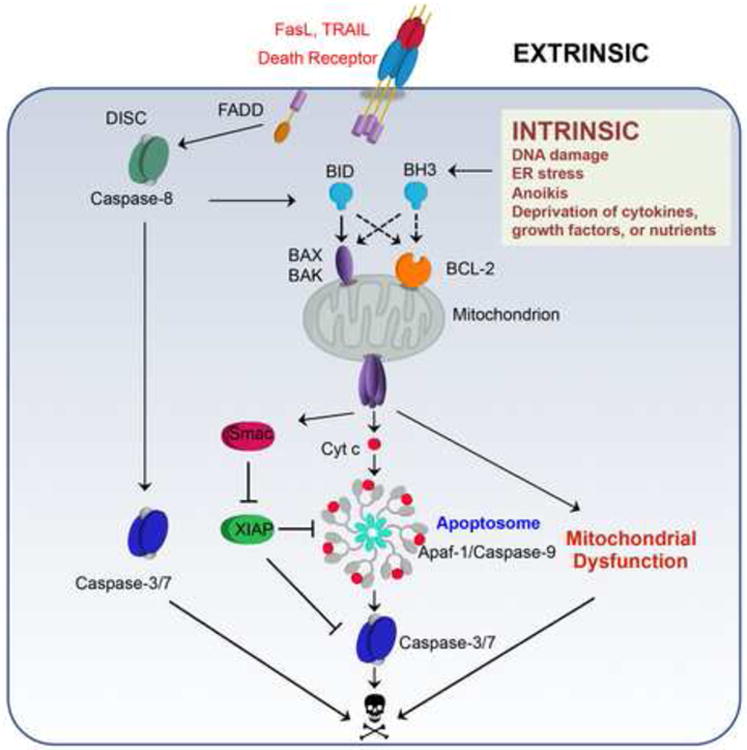
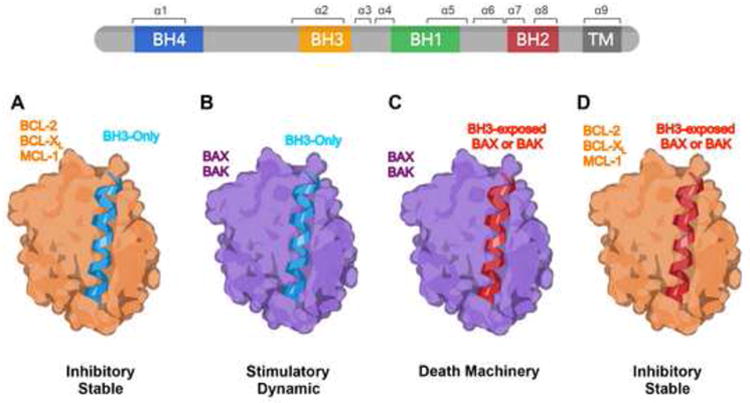
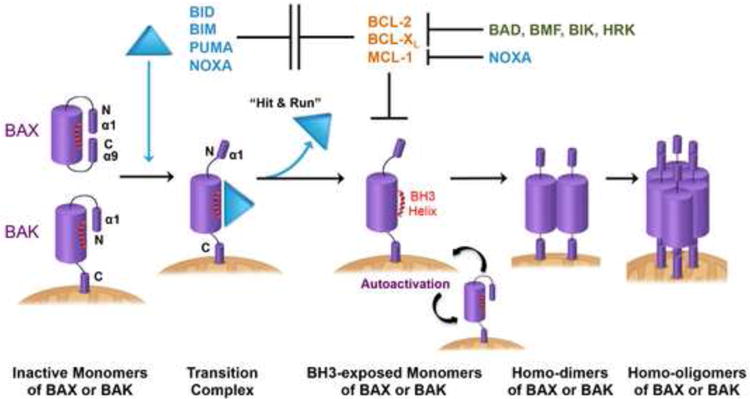
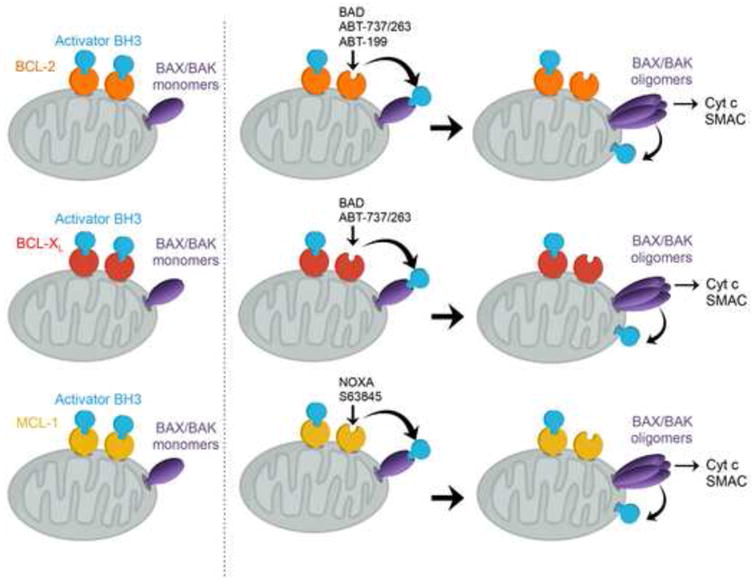
Mitochondria play key roles in mammalian apoptosis, a highly regulated genetic program of cell suicide. Multiple apoptotic signals culminate in mitochondrial outer membrane permeabilization (MOMP), which not only couples the mitochondria to the activation of caspases but also initiates caspase-independent mitochondrial dysfunction. The BCL-2 family proteins are central regulators of MOMP. Multidomain pro-apoptotic BAX and BAK are essential effectors responsible for MOMP, whereas anti-apoptotic BCL-2, BCL-XL, and MCL-1 preserve mitochondrial integrity. The third BCL-2 subfamily of proteins, BH3-only molecules, promotes apoptosis by either activating BAX and BAK or inactivating BCL-2, BCL-XL, and MCL-1. Through an interconnected hierarchical network of interactions, the BCL-2 family proteins integrate developmental and environmental cues to dictate the survival versus death decision of cells by regulating the integrity of the mitochondrial outer membrane. Over the past 30 years, research on the BCL-2-regulated apoptotic pathway has not only revealed its importance in both normal physiological and disease processes, but has also resulted in the first anti-cancer drug targeting protein-protein interactions.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. - PubMed
-
- Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. - PubMed
-
- Korsmeyer SJ. Bcl-2 initiates a new category of oncogenes: regulators of cell death. Blood. 1992;80:879–886. - PubMed
-
- Brem EA, Letai A. BOK: Oddball of the BCL-2 Family. Trends in cell biology. 2016;26:389–390. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous