

Inflammation, Immunity, and Infection in Atherothrombosis: JACC Review Topic of the Week
- PMID: 30336831
- PMCID: PMC6196735
- DOI: 10.1016/j.jacc.2018.08.1043
Inflammation, Immunity, and Infection in Atherothrombosis: JACC Review Topic of the Week
Abstract
Observations on human and experimental atherosclerosis, biomarker studies, and now a large-scale clinical trial support the operation of immune and inflammatory pathways in this disease. The factors that incite innate and adaptive immune responses implicated in atherogenesis and in lesion complication include traditional risk factors such as protein and lipid components of native and modified low-density lipoprotein, angiotensin II, smoking, visceral adipose tissue, and dysmetabolism. Infectious processes and products of the endogenous microbiome might also modulate atherosclerosis and its complications either directly, or indirectly by eliciting local and systemic responses that potentiate disease expression. Trials with antibiotics have not reduced recurrent cardiovascular events, nor have vaccination strategies yet achieved clinical translation. However, anti-inflammatory interventions such as anticytokine therapy and colchicine have begun to show efficacy in this regard. Thus, inflammatory and immune mechanisms can link traditional and emerging risk factors to atherosclerosis, and offer novel avenues for therapeutic intervention.
Keywords: basic & translational research.
Copyright © 2018 American College of Cardiology Foundation. All rights reserved.
Figures

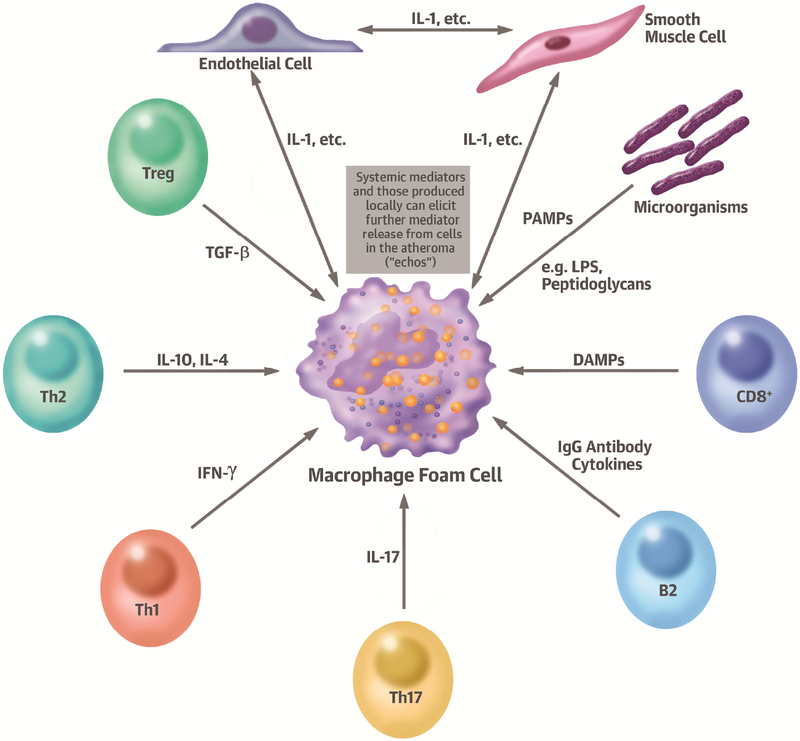
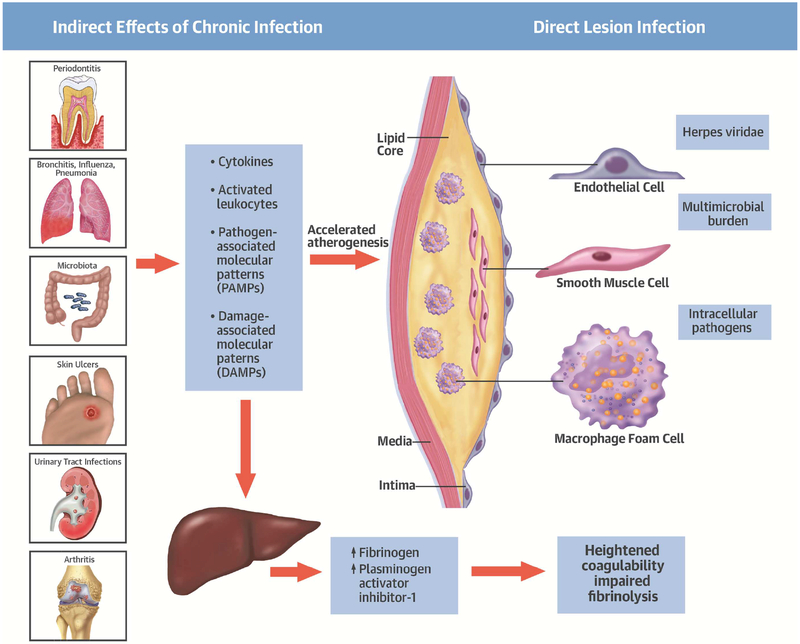
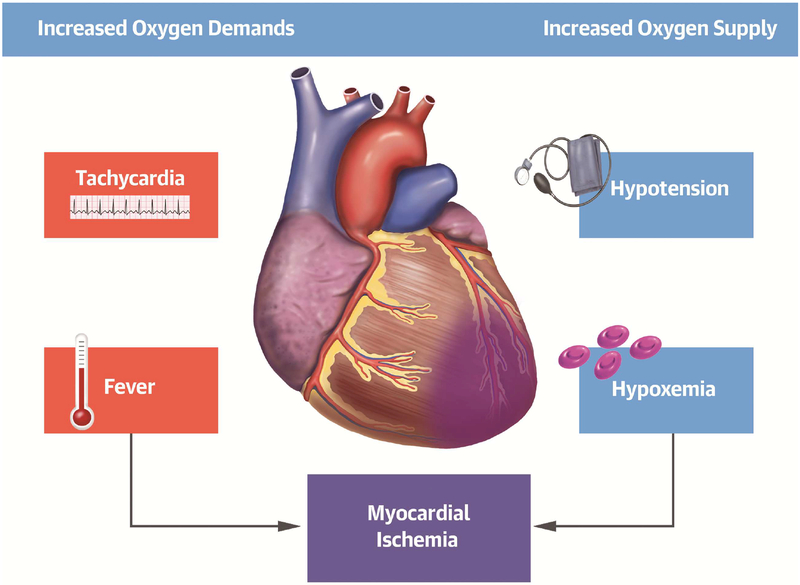
Comment in
-
Inflammation influences thrombus burden in STEMI?Acta Cardiol. 2023 Feb;78(1):164. doi: 10.1080/00015385.2022.2047441. Epub 2022 Mar 3. Acta Cardiol. 2023. PMID: 35240946 No abstract available.
References
-
- Hansson GK, Jonasson L. The discovery of cellular immunity in the atherosclerotic plaque. Arteriosclerosis and Thrombosis: a journal of vascular biology 2009;29:1714–7. - PubMed
-
- Ross R, Harker L. Hyperlipidemia and atherosclerosis. Science 1976;193:1094–100. - PubMed
-
- Ross R, Glomset JA. The pathogenesis of atherosclerosis II. New England Journal of Medicine 1976;295:420–425. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
