

Sotatercept, a novel transforming growth factor β ligand trap, improves anemia in β-thalassemia: a phase II, open-label, dose-finding study
- PMID: 30337358
- PMCID: PMC6395345
- DOI: 10.3324/haematol.2018.198887
Sotatercept, a novel transforming growth factor β ligand trap, improves anemia in β-thalassemia: a phase II, open-label, dose-finding study
Abstract
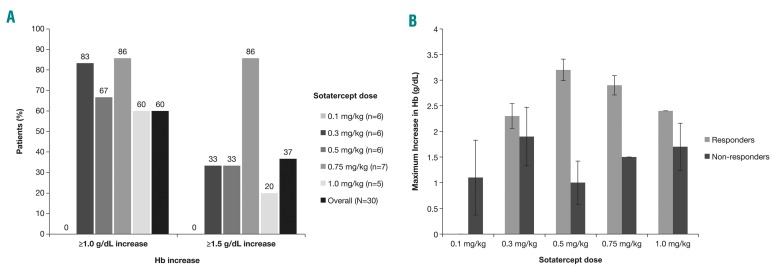
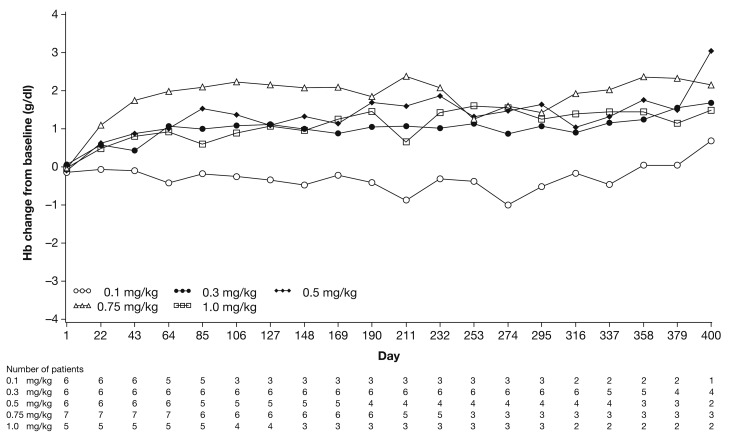
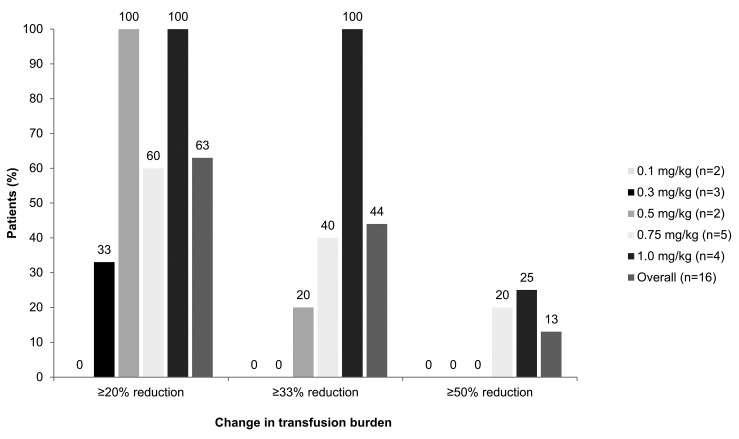
β-thalassemia, a hereditary blood disorder caused by defective synthesis of hemoglobin β globin chains, leads to ineffective erythropoiesis and chronic anemia that may require blood transfusions. Sotatercept (ACE-011) acts as a ligand trap to inhibit negative regulators of late-stage erythropoiesis in the transforming growth factor β superfamily, correcting ineffective erythropoiesis. In this phase II, open-label, dose-finding study, 16 patients with transfusion-dependent β -thalassemia and 30 patients with non-transfusion-dependent β-thalassemia were enrolled at seven centers in four countries between November 2012 and November 2014. Patients were treated with sotatercept at doses of 0.1, 0.3, 0.5, 0.75, or 1.0 mg/kg to determine a safe and effective dose. Doses were administered by subcutaneous injection every 3 weeks. Patients were treated for ≤22 months. Response was assessed as a ≥20% reduction in transfusion burden sustained for 24 weeks in transfusion-dependent β-thalassemia patients, and an increase in hemoglobin level of ≥1.0 g/dL sustained for 12 weeks in non-transfusion-dependent β-thalassemia patients. Sotatercept was well tolerated. After a median treatment duration of 14.4 months (range 0.6-35.9), no severe life-threatening adverse events were observed. Thirteen percent of patients reported serious but manageable adverse events. The active dose of sotatercept was ≥0.3 mg/kg for patients with non-transfusion-dependent β-thalassemia and ≥0.5 mg/kg for those with transfusion-dependent β-thalassemia. Of 30 non-transfusion-dependent β-thalassemia patients treated with ≥0.1 mg/kg sotatercept, 18 (60%) achieved a mean hemoglobin increase ≥1.0 g/dL, and 11 (37%) an increase ≥1.5 g/dL, sustained for ≥12 weeks. Four (100%) transfusion-dependent β-thalassemia patients treated with 1.0 mg/kg sotatercept achieved a transfusion-burden reduction of ≥20%. Sotatercept was effective and well tolerated in patients with β-thalassemia. Most patients with non-transfusion-dependent β-thalassemia treated with higher doses achieved sustained increases in hemoglobin level. Transfusion-dependent β-thalassemia patients treated with higher doses of sotatercept achieved notable reductions in transfusion requirements. This trial was registered at ClinicalTrials.gov with the number NCT01571635.
Copyright© 2019 Ferrata Storti Foundation.
Figures
References
-
- Malamos B, Belcher EH, Gyftaki E, Binopoulos D. Simultaneous studies with Fe59 and Cr51 in congenital haemolytic anaemias. Nucl Med (Stuttg). 1961;2:1–20. - PubMed
-
- Weatherall DJ, Clegg JB. The Thalassemia Syndromes. 2nd ed. Oxford, England: Blackwell Scientific Publications; 1972.
-
- Arlet JB, Dussiot M, Moura IC, Hermine O, Courtois G. Novel players in β-thalassemia dyserythropoiesis and new therapeutic strategies. Curr Opin Hematol. 2016;23(3): 181–188. - PubMed
-
- Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005;353(11):1135–1146. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
