

The molecular basis of protein toxin HicA-dependent binding of the protein antitoxin HicB to DNA
- PMID: 30337369
- PMCID: PMC6302177
- DOI: 10.1074/jbc.RA118.005173
The molecular basis of protein toxin HicA-dependent binding of the protein antitoxin HicB to DNA
Abstract
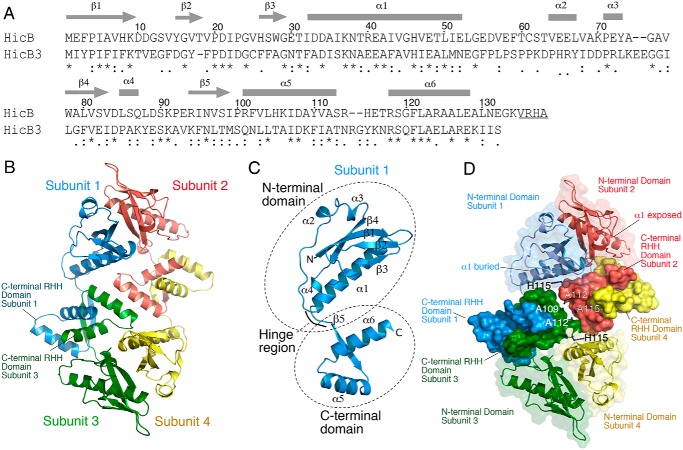
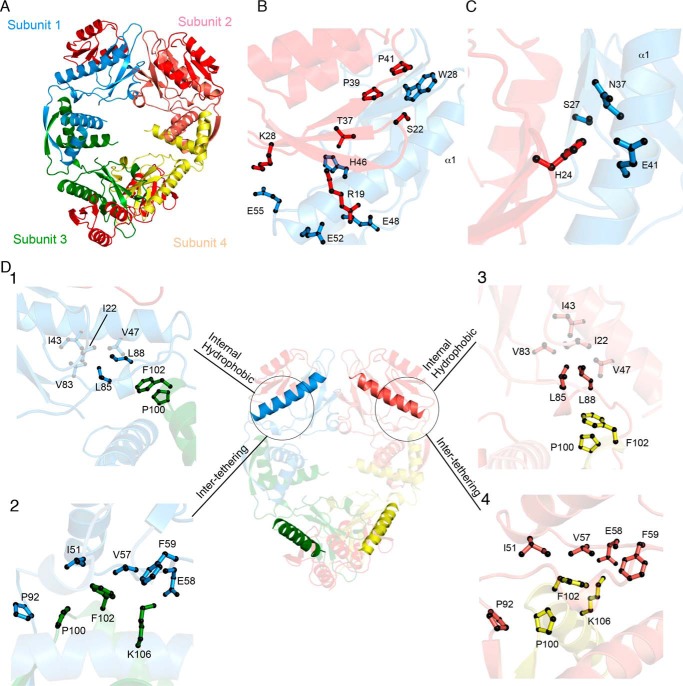
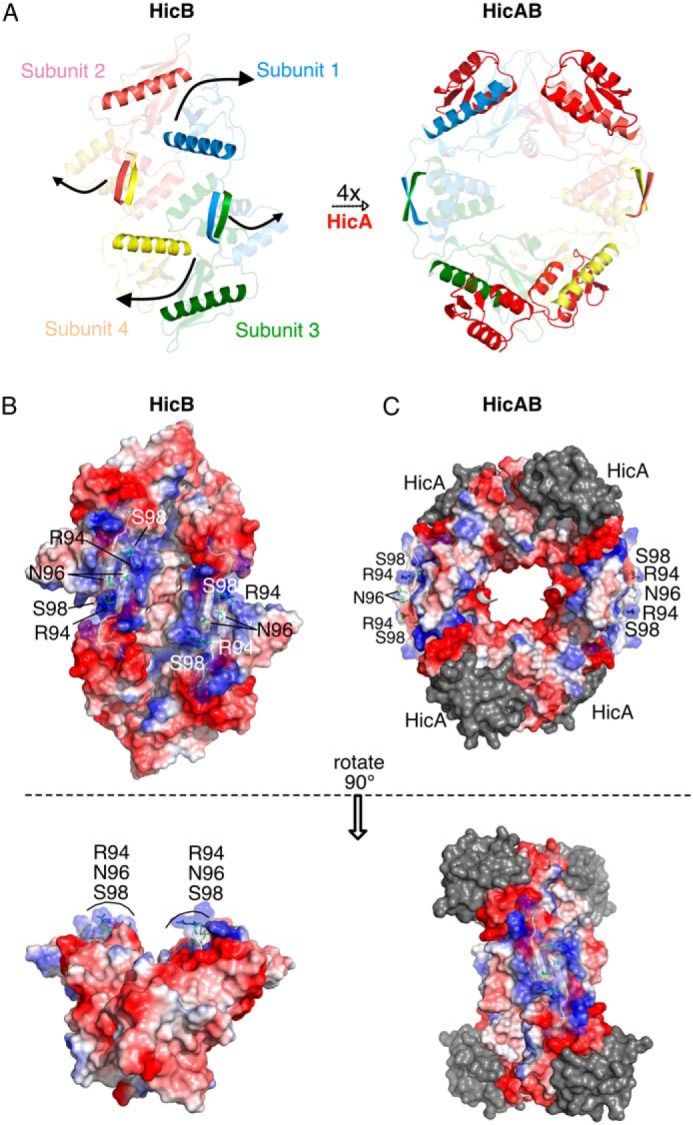
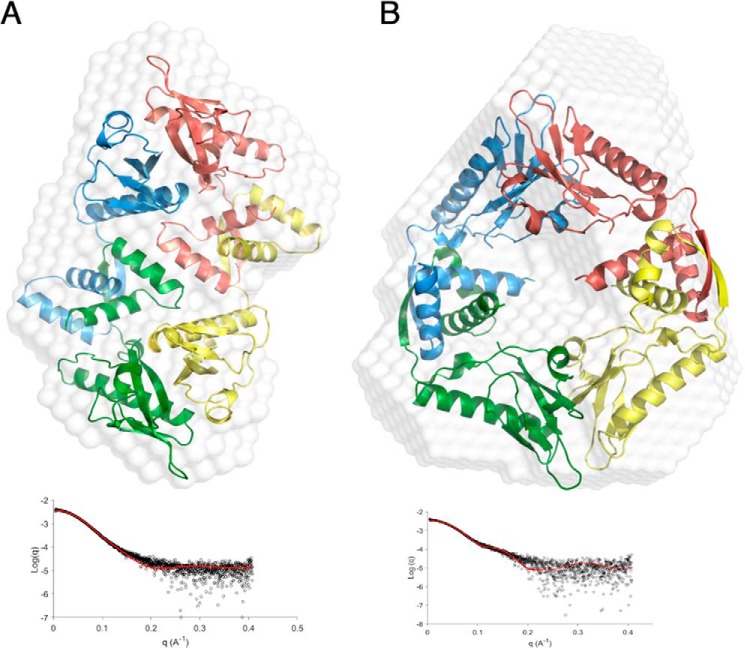
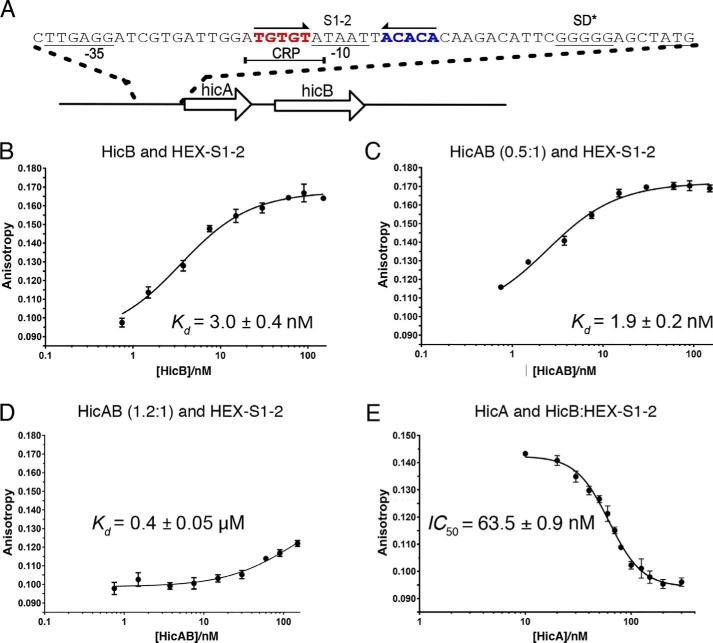
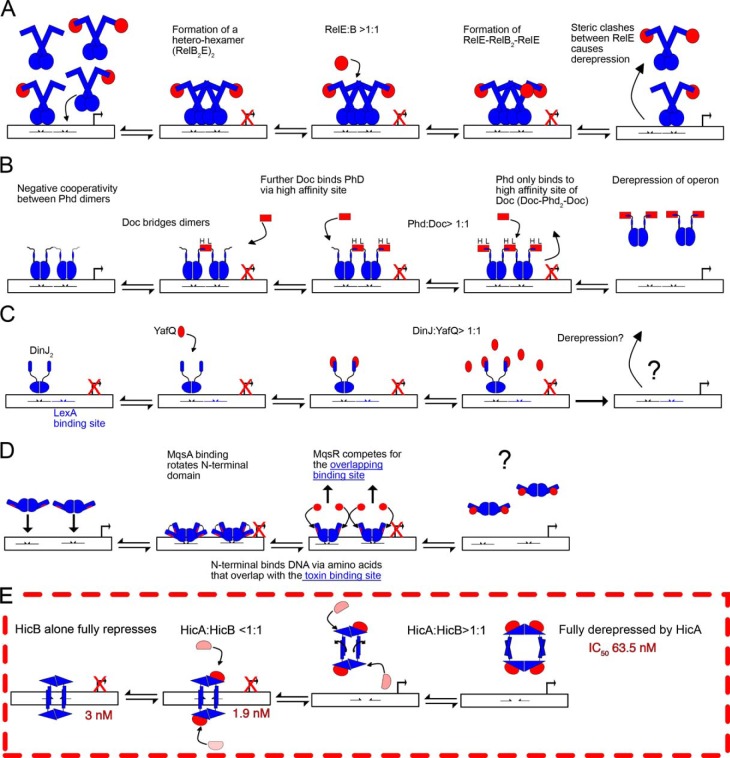
Toxin-antitoxin (TA) systems are present in many bacteria and play important roles in bacterial growth, physiology, and pathogenicity. Those that are best studied are the type II TA systems, in which both toxins and antitoxins are proteins. The HicAB system is one of the prototypic TA systems, found in many bacterial species. Complex interactions between the protein toxin (HicA), the protein antitoxin (HicB), and the DNA upstream of the encoding genes regulate the activity of this system, but few structural details are available about how HicA destabilizes the HicB-DNA complex. Here, we determined the X-ray structures of HicB and the HicAB complex to 1.8 and 2.5 Å resolution, respectively, and characterized their DNA interactions. This revealed that HicB forms a tetramer and HicA and HicB form a heterooctameric complex that involves structural reorganization of the C-terminal (DNA-binding) region of HicB. Our observations indicated that HicA has a profound impact on binding of HicB to DNA sequences upstream of hicAB in a stoichiometric-dependent way. At low ratios of HicA:HicB, there was no effect on DNA binding, but at higher ratios, the affinity for DNA declined cooperatively, driving dissociation of the HicA:HicB:DNA complex. These results reveal the structural mechanisms by which HicA de-represses the HicB-DNA complex.
Keywords: DNA-binding protein; HicAB; X-ray crystallography; antibiotic resistance; bacterial toxin; conditional cooperativity; persistence; protein-protein interaction; structural biology; toxin–antitoxin system; type II TA system.
© 2018 Winter et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
Similar articles
-
The HicAB System: Characteristics and Biological Roles of an Underappreciated Toxin-Antitoxin System.Int J Mol Sci. 2024 Nov 13;25(22):12165. doi: 10.3390/ijms252212165. Int J Mol Sci. 2024. PMID: 39596231 Free PMC article. Review.
-
HicA toxin of Escherichia coli derepresses hicAB transcription to selectively produce HicB antitoxin.Mol Microbiol. 2017 Jun;104(5):781-792. doi: 10.1111/mmi.13662. Epub 2017 Mar 21. Mol Microbiol. 2017. PMID: 28266056
-
Characterization of HicAB toxin-antitoxin module of Sinorhizobium meliloti.BMC Microbiol. 2019 Jan 10;19(1):10. doi: 10.1186/s12866-018-1382-6. BMC Microbiol. 2019. PMID: 30630415 Free PMC article.
-
Activation of Toxin-Antitoxin System Toxins Suppresses Lethality Caused by the Loss of σE in Escherichia coli.J Bacteriol. 2015 Jul;197(14):2316-24. doi: 10.1128/JB.00079-15. Epub 2015 Apr 27. J Bacteriol. 2015. PMID: 25917909 Free PMC article.
-
Conformational change as a mechanism for toxin activation in bacterial toxin-antitoxin systems.J Virol. 2024 Nov 19;98(11):e0151324. doi: 10.1128/jvi.01513-24. Epub 2024 Oct 24. J Virol. 2024. PMID: 39445801 Free PMC article. Review.
Cited by
-
Diverse genetic contexts of HicA toxin domains propose a role in anti-phage defense.mBio. 2024 Feb 14;15(2):e0329323. doi: 10.1128/mbio.03293-23. Epub 2024 Jan 18. mBio. 2024. PMID: 38236063 Free PMC article.
-
The HicAB System: Characteristics and Biological Roles of an Underappreciated Toxin-Antitoxin System.Int J Mol Sci. 2024 Nov 13;25(22):12165. doi: 10.3390/ijms252212165. Int J Mol Sci. 2024. PMID: 39596231 Free PMC article. Review.
-
Dynamics-Based Regulatory Switches of Type II Antitoxins: Insights into New Antimicrobial Discovery.Antibiotics (Basel). 2023 Mar 23;12(4):637. doi: 10.3390/antibiotics12040637. Antibiotics (Basel). 2023. PMID: 37106997 Free PMC article.
-
Type II Toxin-Antitoxin Systems in Escherichia coli.Infect Drug Resist. 2025 Feb 24;18:1083-1096. doi: 10.2147/IDR.S501485. eCollection 2025. Infect Drug Resist. 2025. PMID: 40027916 Free PMC article. Review.
-
Prokaryote toxin-antitoxin modules: Complex regulation of an unclear function.Protein Sci. 2021 Jun;30(6):1103-1113. doi: 10.1002/pro.4071. Epub 2021 Apr 7. Protein Sci. 2021. PMID: 33786944 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
