

E-cadherin loss induces targetable autocrine activation of growth factor signalling in lobular breast cancer
- PMID: 30337563
- PMCID: PMC6193986
- DOI: 10.1038/s41598-018-33525-5
E-cadherin loss induces targetable autocrine activation of growth factor signalling in lobular breast cancer
Abstract
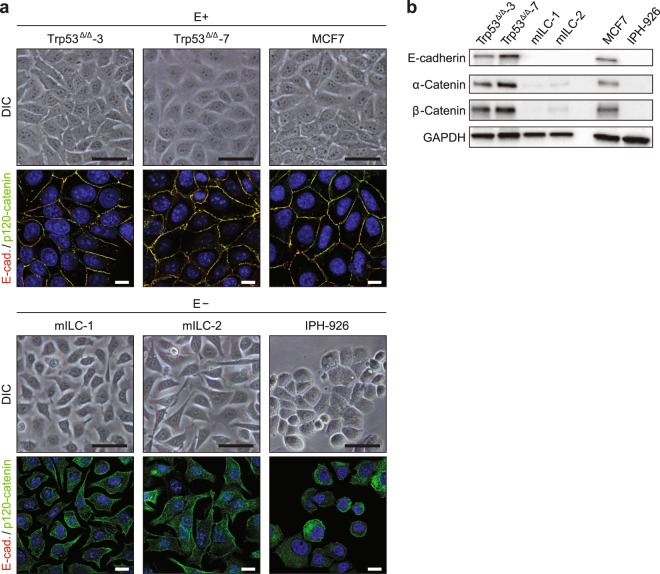
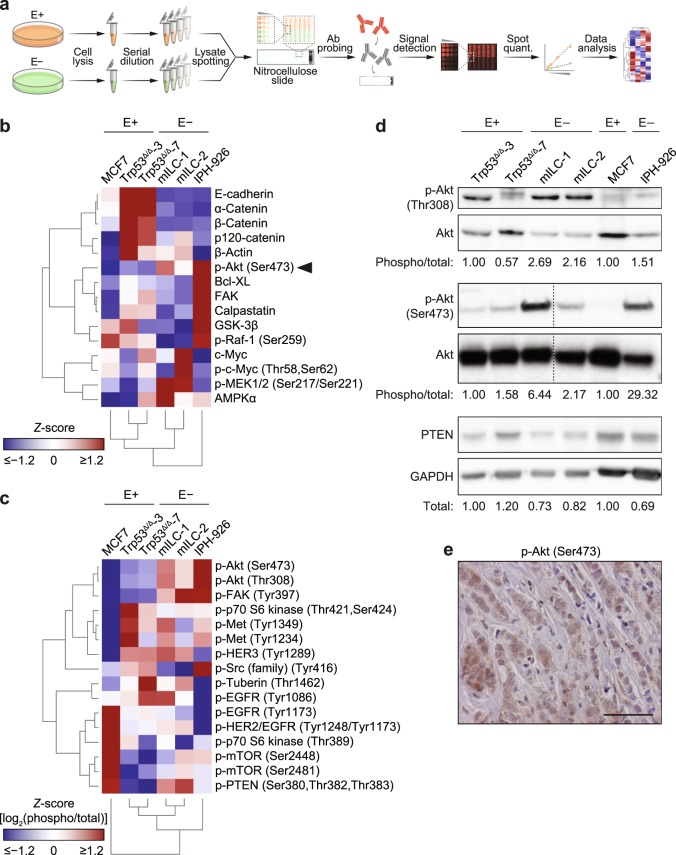
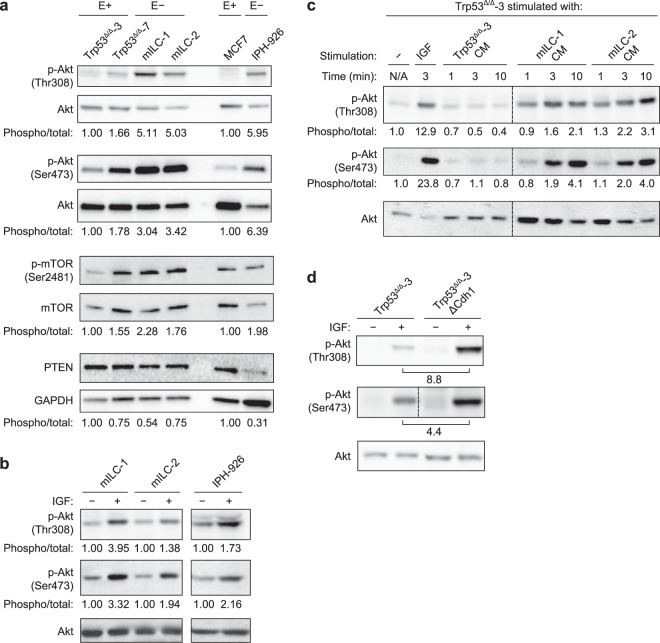
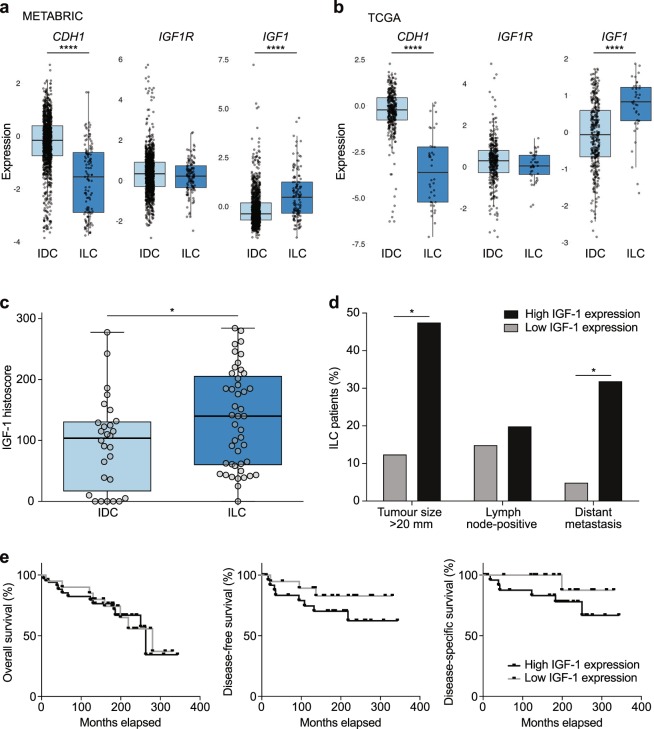
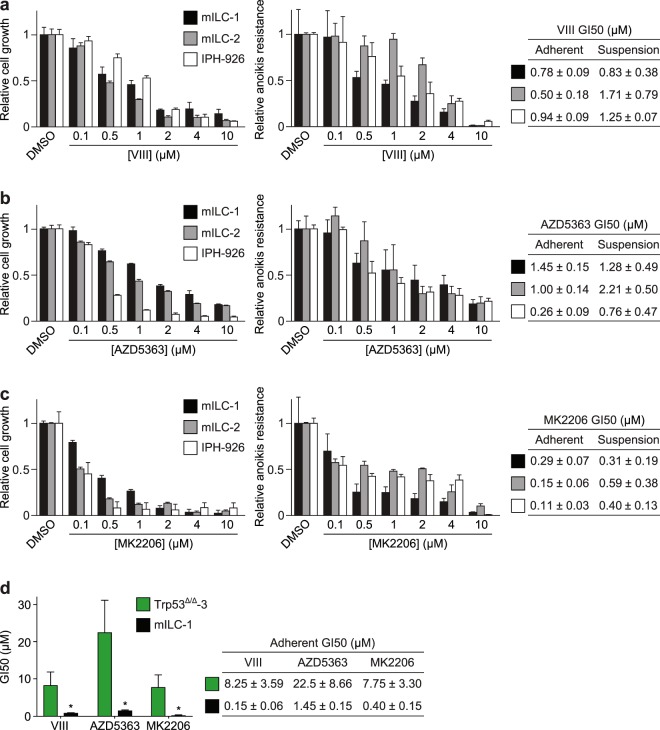
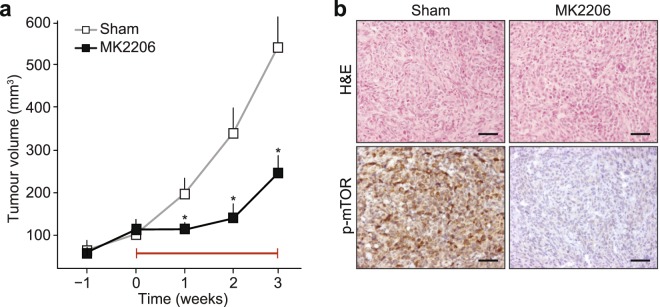
Despite the fact that loss of E-cadherin is causal to the development and progression of invasive lobular carcinoma (ILC), options to treat this major breast cancer subtype are limited if tumours develop resistance to anti-oestrogen treatment regimens. This study aimed to identify clinically targetable pathways that are aberrantly active downstream of E-cadherin loss in ILC. Using a combination of reverse-phase protein array (RPPA) analyses, mRNA sequencing, conditioned medium growth assays and CRISPR/Cas9-based knock-out experiments, we demonstrate that E-cadherin loss causes increased responsiveness to autocrine growth factor receptor (GFR)-dependent activation of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/Akt signalling. Autocrine activation of GFR signalling and its downstream PI3K/Akt hub was independent of oncogenic mutations in PIK3CA, AKT1 or PTEN. Analyses of human ILC samples confirmed growth factor production and pathway activity. Pharmacological inhibition of Akt using AZD5363 or MK2206 resulted in robust inhibition of cell growth and survival of ILC cells, and impeded tumour growth in a mouse ILC model. Because E-cadherin loss evokes hypersensitisation of PI3K/Akt activation independent of oncogenic mutations in this pathway, we propose clinical intervention of PI3K/Akt in ILC based on functional E-cadherin inactivation, irrespective of activating pathway mutations.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- KWF-UU-10456/KWF Kankerbestrijding (Dutch Cancer Society)/International
- C157/A15703/Cancer Research UK (CRUK)/International
- KWF-UU-2011-5230/KWF Kankerbestrijding (Dutch Cancer Society)/International
- NWO/ZonMW-VIDI 0616.096.318/Netherlands Organisation for Scientific Research | Stichting voor de Technische Wetenschappen (Technology Foundation STW)/International
- Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
