

Crystallographic snapshots of ligand binding to hexameric purine nucleoside phosphorylase and kinetic studies give insight into the mechanism of catalysis
- PMID: 30337572
- PMCID: PMC6193948
- DOI: 10.1038/s41598-018-33723-1
Crystallographic snapshots of ligand binding to hexameric purine nucleoside phosphorylase and kinetic studies give insight into the mechanism of catalysis
Abstract
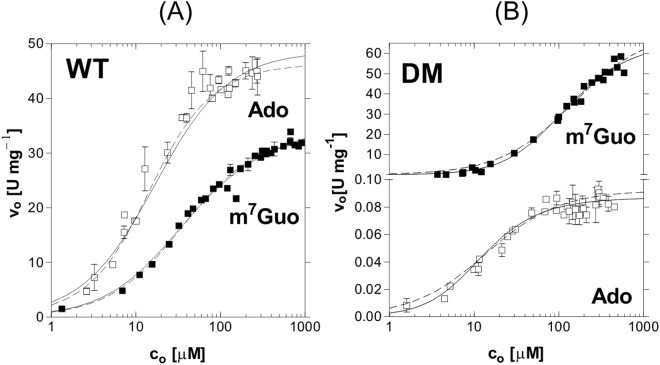
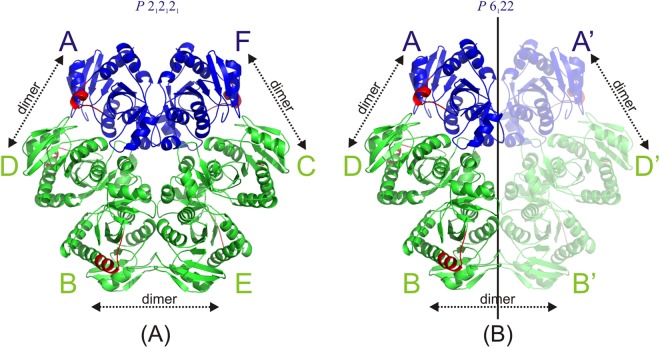
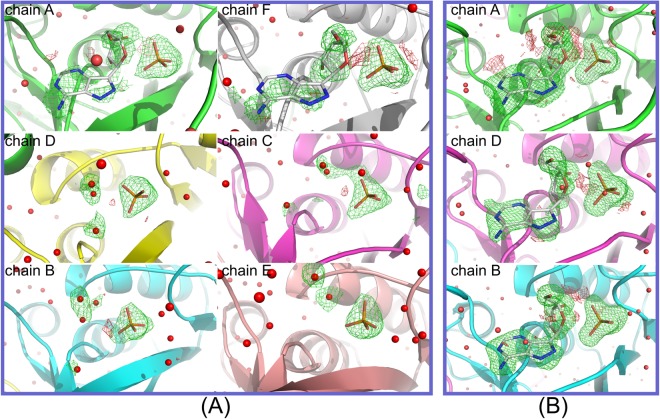
Purine nucleoside phosphorylase (PNP) catalyses the cleavage of the glycosidic bond of purine nucleosides using phosphate instead of water as a second substrate. PNP from Escherichia coli is a homohexamer, build as a trimer of dimers, and each subunit can be in two conformations, open or closed. This conformational change is induced by the presence of phosphate substrate, and very likely a required step for the catalysis. Closing one active site strongly affects the others, by a yet unclear mechanism and order of events. Kinetic and ligand binding studies show strong negative cooperativity between subunits. Here, for the first time, we managed to monitor the sequence of nucleoside binding to individual subunits in the crystal structures of the wild-type enzyme, showing that first the closed sites, not the open ones, are occupied by the nucleoside. However, two mutations within the active site, Asp204Ala/Arg217Ala, are enough not only to significantly reduce the effectiveness of the enzyme, but also reverse the sequence of the nucleoside binding. In the mutant the open sites, neighbours in a dimer of those in the closed conformation, are occupied as first. This demonstrates how important for the effective catalysis of Escherichia coli PNP is proper subunit cooperation.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Štefanić Z, et al. Still a Long Way to Fully Understanding the Molecular Mechanism of Escherichia coli Purine Nucleoside Phosphorylase. Croat. Chem. Acta. 2013;86:117–127. doi: 10.5562/cca2116. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
