

Genomic analysis of Acinetobacter baumannii prophages reveals remarkable diversity and suggests profound impact on bacterial virulence and fitness
- PMID: 30337588
- PMCID: PMC6193963
- DOI: 10.1038/s41598-018-33800-5
Genomic analysis of Acinetobacter baumannii prophages reveals remarkable diversity and suggests profound impact on bacterial virulence and fitness
Abstract
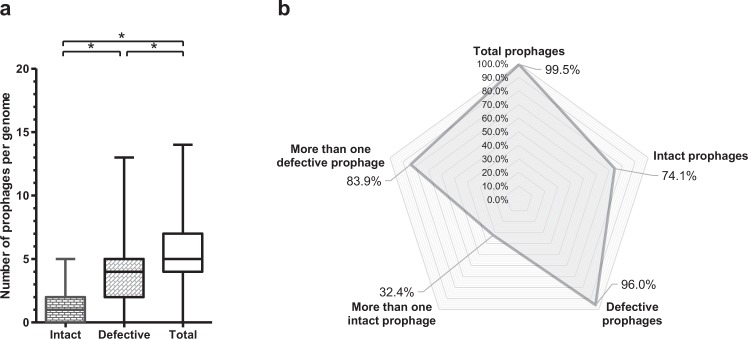
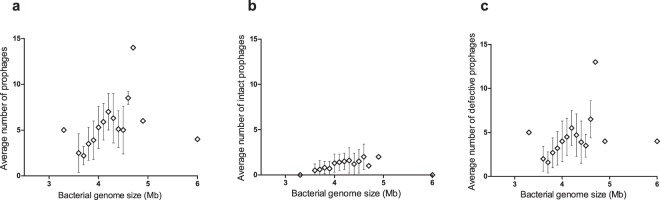
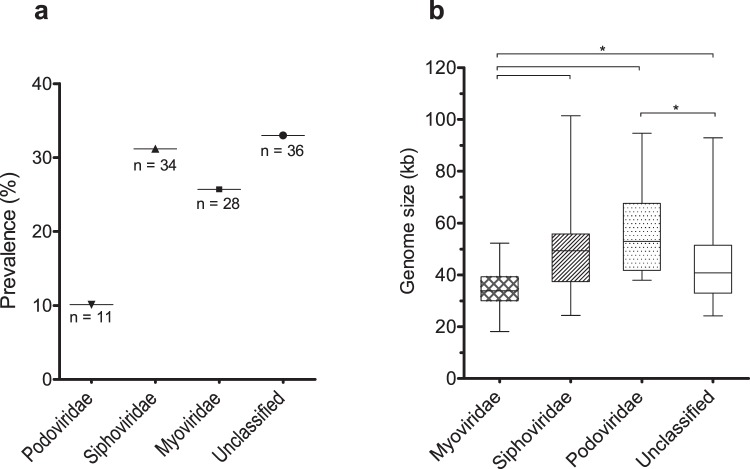
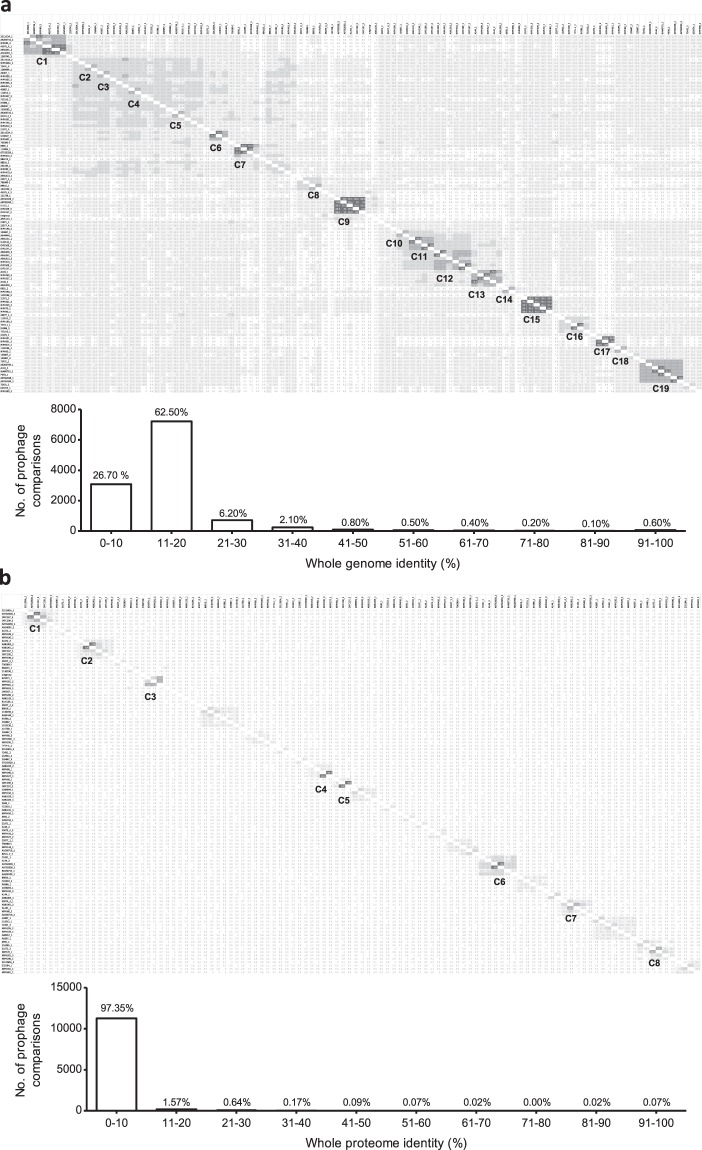
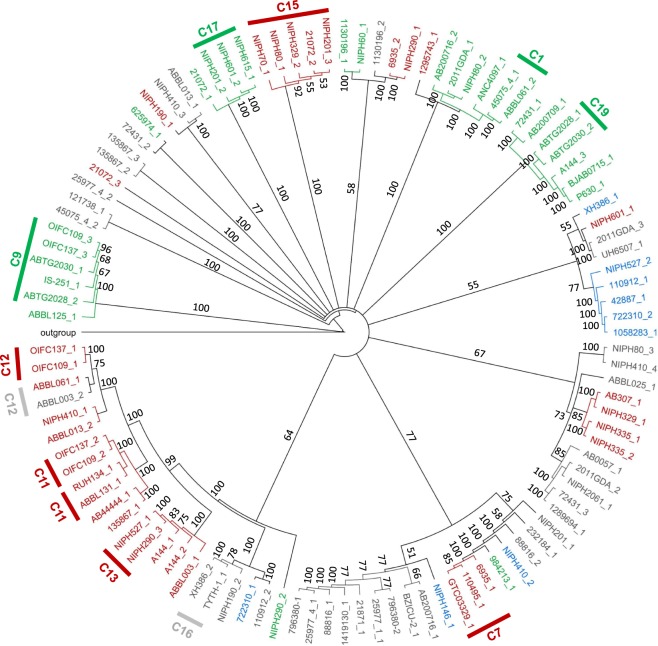
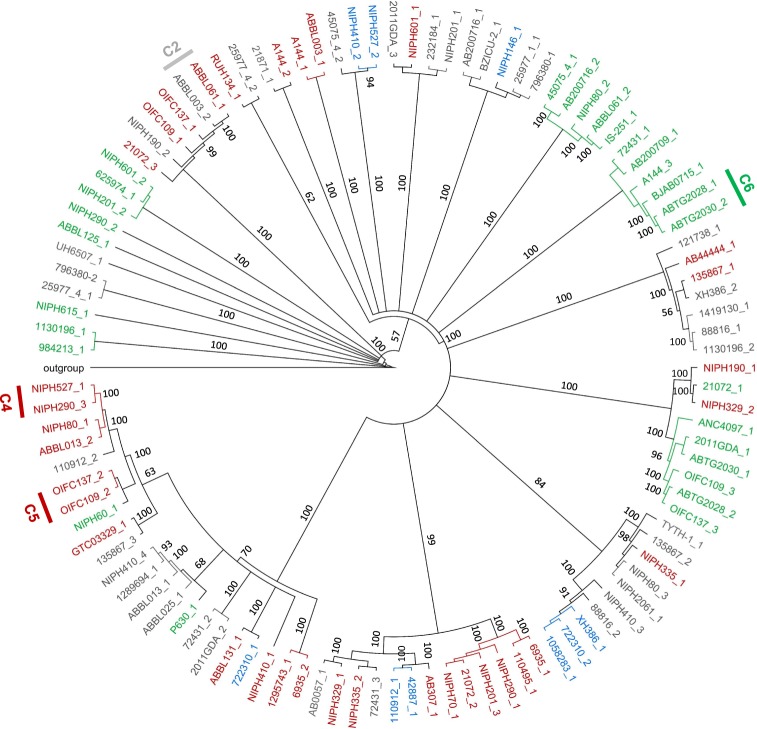
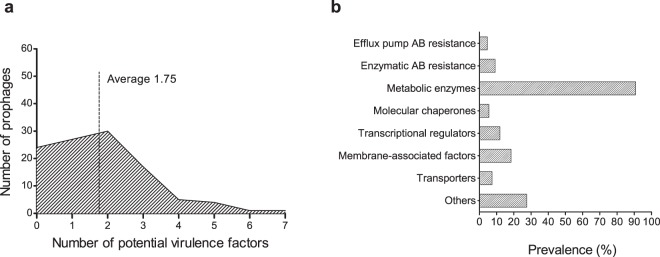
The recent nomination by the World Health Organization of Acinetobacter baumannii as the number one priority pathogen for the development of new antibiotics is a direct consequence of its fast evolution of pathogenicity, and in particular of multidrug resistance. While the development of new antibiotics is critical, understanding the mechanisms behind the crescent bacterial antibiotic resistance is equally relevant. Often, resistance and other bacterial virulence elements are contained on highly mobile pieces of DNA that can easily spread to other bacteria. Prophages are one of the mediators of this form of gene transfer, and have been frequently found in bacterial genomes, often offering advantageous features to the host. Here we assess the contribution of prophages for the evolution of A. baumannii pathogenicity. We found prophages to be notably diverse and widely disseminated in A. baumannii genomes. Also remarkably, A. baumannii prophages encode for multiple putative virulence factors that may be implicated in the bacterium's capacity to colonize host niches, evade the host immune system, subsist in unfavorable environments, and tolerate antibiotics. Overall our results point towards a significant contribution of prophages for the dissemination and evolution of pathogenicity in A. baumannii, and highlight their clinical relevance.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- SFRH/BPD/94648/2013/Ministry of Education and Science | Fundação para a Ciência e a Tecnologia (Portuguese Science and Technology Foundation)/International
- PTDC/BBB-BSS/6471/2014/Ministry of Education and Science | Fundação para a Ciência e a Tecnologia (Portuguese Science and Technology Foundation)/International
- UID/BIO/04469/2013/Ministry of Education and Science | Fundação para a Ciência e a Tecnologia (Portuguese Science and Technology Foundation)/International
- POCI-01-0145-FEDER-006684/Ministry of Education and Science | Fundação para a Ciência e a Tecnologia (Portuguese Science and Technology Foundation)/International
- PTDC/BBB-BSS/6471/2014/Ministry of Education and Science | Fundação para a Ciência e a Tecnologia (Portuguese Science and Technology Foundation)/International
- UID/BIO/04469/2013/Ministry of Education and Science | Fundação para a Ciência e a Tecnologia (Portuguese Science and Technology Foundation)/International
- POCI-01-0145-FEDER-006684/Ministry of Education and Science | Fundação para a Ciência e a Tecnologia (Portuguese Science and Technology Foundation)/International
- PTDC/BBB-BSS/6471/2014/Ministry of Education and Science | Fundação para a Ciência e a Tecnologia (Portuguese Science and Technology Foundation)/International
- POCI-01-0145-FEDER-006684/Ministry of Education and Science | Fundação para a Ciência e a Tecnologia (Portuguese Science and Technology Foundation)/International
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
