

Treating oxidative stress in heart failure: past, present and future
- PMID: 30338885
- PMCID: PMC6607515
- DOI: 10.1002/ejhf.1320
Treating oxidative stress in heart failure: past, present and future
Abstract
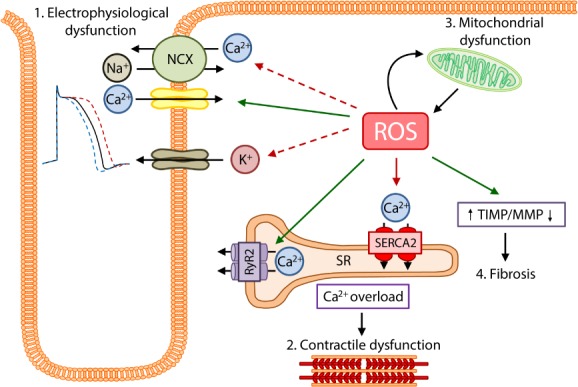
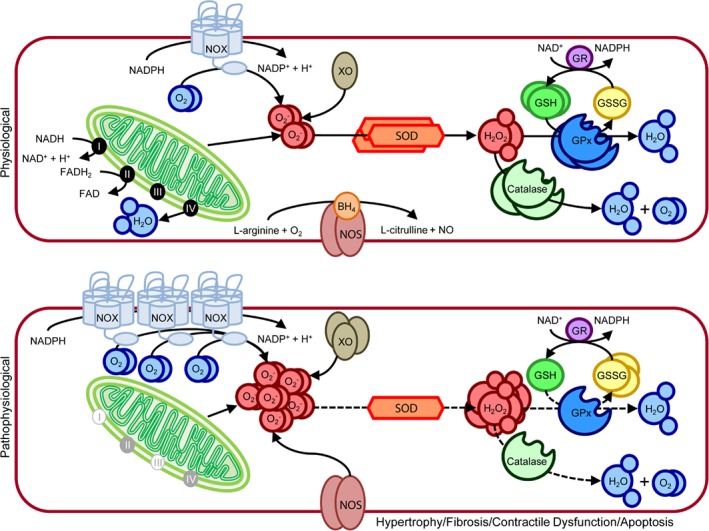
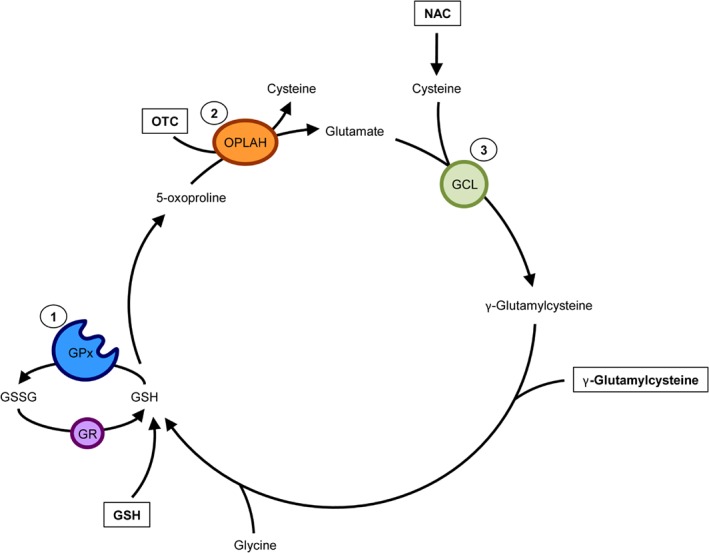
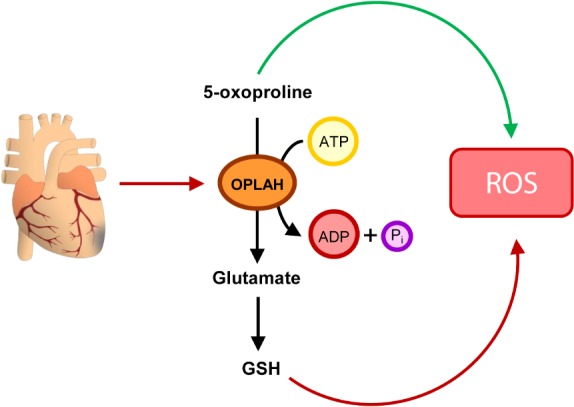
Advances in cardiovascular research have identified oxidative stress as an important pathophysiological pathway in the development and progression of heart failure. Oxidative stress is defined as the imbalance between the production of reactive oxygen species (ROS) and the endogenous antioxidant defence system. Under physiological conditions, small quantities of ROS are produced intracellularly, which function in cell signalling, and can be readily reduced by the antioxidant defence system. However, under pathophysiological conditions, the production of ROS exceeds the buffering capacity of the antioxidant defence system, resulting in cell damage and death. Over the last decades several studies have tried to target oxidative stress with the aim to improve outcome in patients with heart failure, with very limited success. The reasons as to why these studies failed to demonstrate any beneficial effects remain unclear. However, one plausible explanation might be that currently employed strategies, which target oxidative stress by exogenous inhibition of ROS production or supplementation of exogenous antioxidants, are not effective enough, while bolstering the endogenous antioxidant capacity might be a far more potent avenue for therapeutic intervention. In this review, we provide an overview of oxidative stress in the pathophysiology of heart failure and the strategies utilized to date to target this pathway. We provide novel insights into modulation of endogenous antioxidants, which may lead to novel therapeutic strategies to improve outcome in patients with heart failure.
Keywords: Glutathione; Heart failure; Nicotinamide adenine dinucleotide; Oxidative stress; γ-Glutamyl cycle.
© 2018 The Authors. European Journal of Heart Failure published by John Wiley & Sons Ltd on behalf of European Society of Cardiology.
Figures
References
-
- Karimi Galougahi K, Antoniades C, Nicholls SJ, Channon KM, Figtree GA. Redox biomarkers in cardiovascular medicine. Eur Heart J 2015;36:1576–1582. - PubMed
-
- Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 2011;301:H2181‐2190. - PubMed
-
- Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P. 2016. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) . Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016;18:891–975. - PubMed
-
- Wolfram R, Oguogho A, Palumbo B, Sinzinger H. Enhanced oxidative stress in coronary heart disease and chronic heart failure as indicated by an increased 8‐epi‐PGF 2α. Eur J Heart Fail 2005;7:167–172. - PubMed
-
- Sato Y, Fujiwara H, Takatsu Y. Cardiac troponin and heart failure in the era of high‐sensitivity assays. J Cardiol 2012;60:160–167. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
