

Large-scale genome-wide enrichment analyses identify new trait-associated genes and pathways across 31 human phenotypes
- PMID: 30341297
- PMCID: PMC6195536
- DOI: 10.1038/s41467-018-06805-x
Large-scale genome-wide enrichment analyses identify new trait-associated genes and pathways across 31 human phenotypes
Abstract
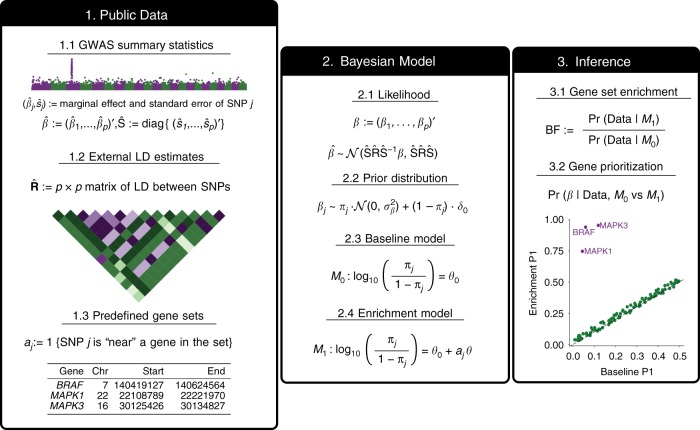
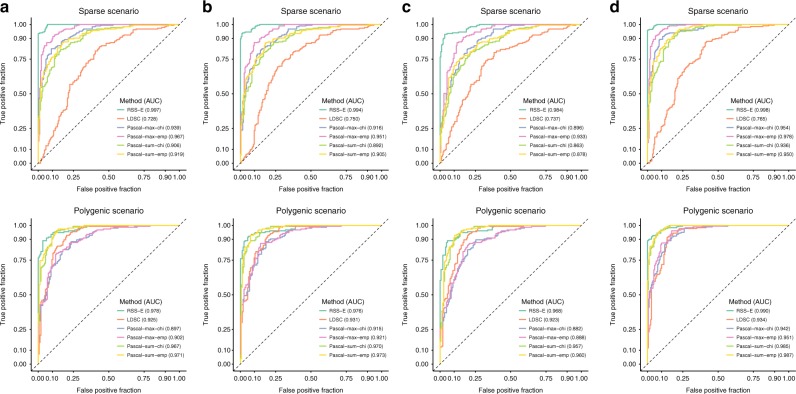
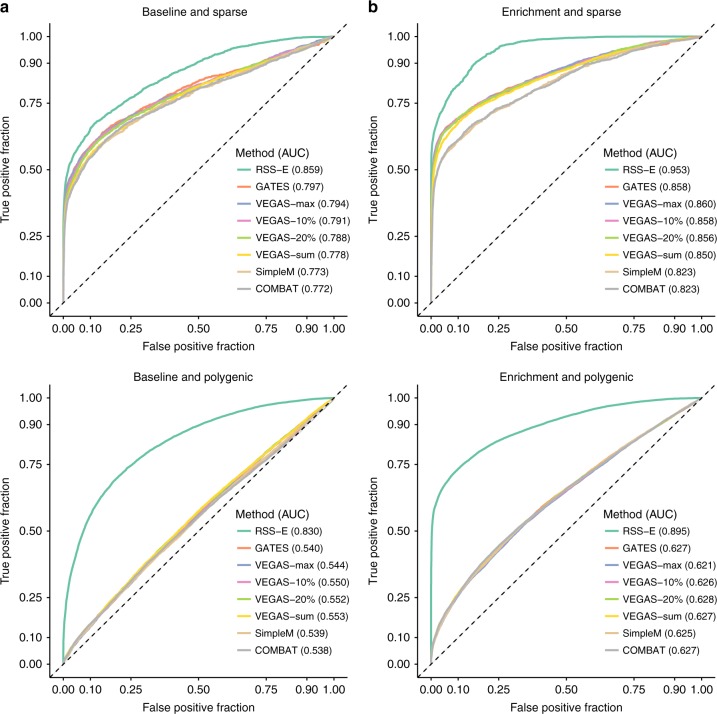
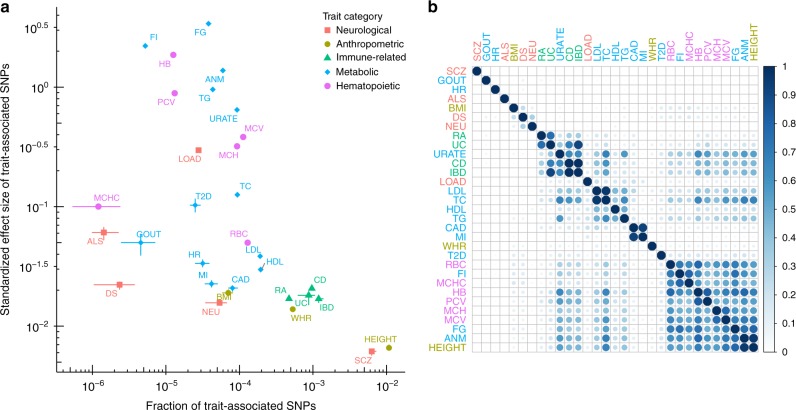
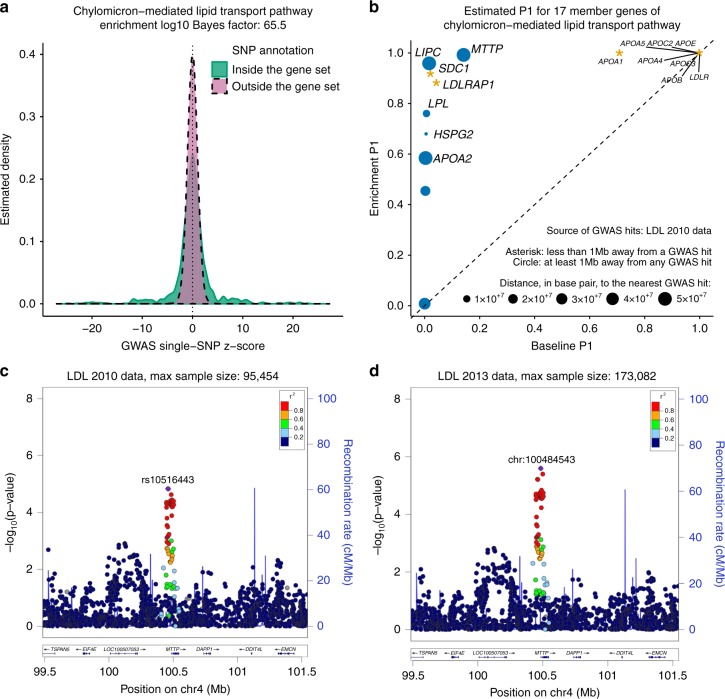
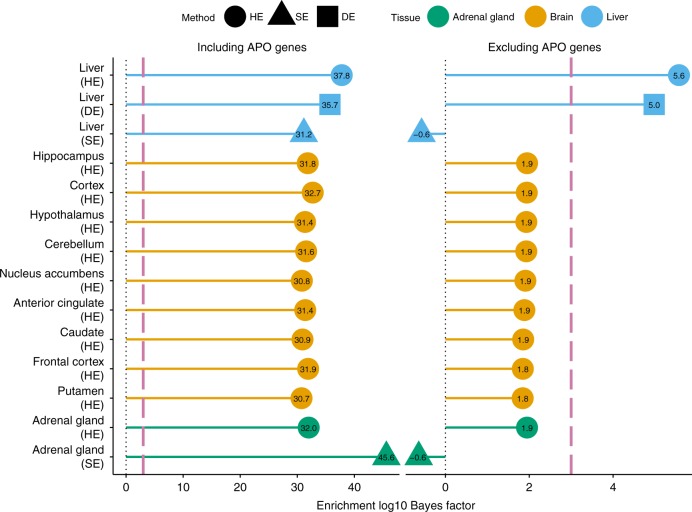
Genome-wide association studies (GWAS) aim to identify genetic factors associated with phenotypes. Standard analyses test variants for associations individually. However, variant-level associations are hard to identify and can be difficult to interpret biologically. Enrichment analyses help address both problems by targeting sets of biologically related variants. Here we introduce a new model-based enrichment method that requires only GWAS summary statistics. Applying this method to interrogate 4,026 gene sets in 31 human phenotypes identifies many previously-unreported enrichments, including enrichments of endochondral ossification pathway for height, NFAT-dependent transcription pathway for rheumatoid arthritis, brain-related genes for coronary artery disease, and liver-related genes for Alzheimer's disease. A key feature of our method is that inferred enrichments automatically help identify new trait-associated genes. For example, accounting for enrichment in lipid transport genes highlights association between MTTP and low-density lipoprotein levels, whereas conventional analyses of the same data found no significant variants near this gene.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
