

Myotonic Dystrophies: Targeting Therapies for Multisystem Disease
- PMID: 30341596
- PMCID: PMC6277298
- DOI: 10.1007/s13311-018-00679-z
Myotonic Dystrophies: Targeting Therapies for Multisystem Disease
Abstract
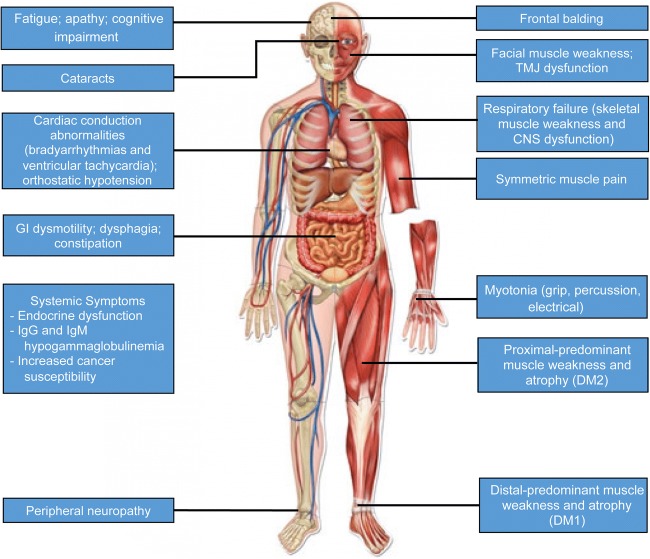
Myotonic dystrophy is an autosomal dominant muscular dystrophy not only associated with muscle weakness, atrophy, and myotonia but also prominent multisystem involvement. There are 2 similar, but distinct, forms of myotonic dystrophy; type 1 is caused by a CTG repeat expansion in the DMPK gene, and type 2 is caused by a CCTG repeat expansion in the CNBP gene. Type 1 is associated with distal limb, neck flexor, and bulbar weakness and results in different phenotypic subtypes with variable onset from congenital to very late-onset as well as variable signs and symptoms. The classically described adult-onset form is the most common. In contrast, myotonic dystrophy type 2 is adult-onset or late-onset, has proximal predominant muscle weakness, and generally has less severe multisystem involvement. In both forms of myotonic dystrophy, the best characterized disease mechanism is a RNA toxic gain-of-function during which RNA repeats form nuclear foci resulting in sequestration of RNA-binding proteins and, therefore, dysregulated splicing of premessenger RNA. There are currently no disease-modifying therapies, but clinical surveillance, preventative measures, and supportive treatments are used to reduce the impact of muscular impairment and other systemic involvement including cataracts, cardiac conduction abnormalities, fatigue, central nervous system dysfunction, respiratory weakness, dysphagia, and endocrine dysfunction. Exciting preclinical progress has been made in identifying a number of potential strategies including genome editing, small molecule therapeutics, and antisense oligonucleotide-based therapies to target the pathogenesis of type 1 and type 2 myotonic dystrophies at the DNA, RNA, or downstream target level.
Keywords: Myotonic dystrophy; biomarker; myopathies; splicing; therapeutic..
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
