

Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk
- PMID: 30343897
- PMCID: PMC6269166
- DOI: 10.1016/j.cell.2018.09.049
Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk
Erratum in
-
Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk.Cell. 2019 Jun 27;178(1):262. doi: 10.1016/j.cell.2019.06.016. Cell. 2019. PMID: 31251915 Free PMC article. No abstract available.
Abstract
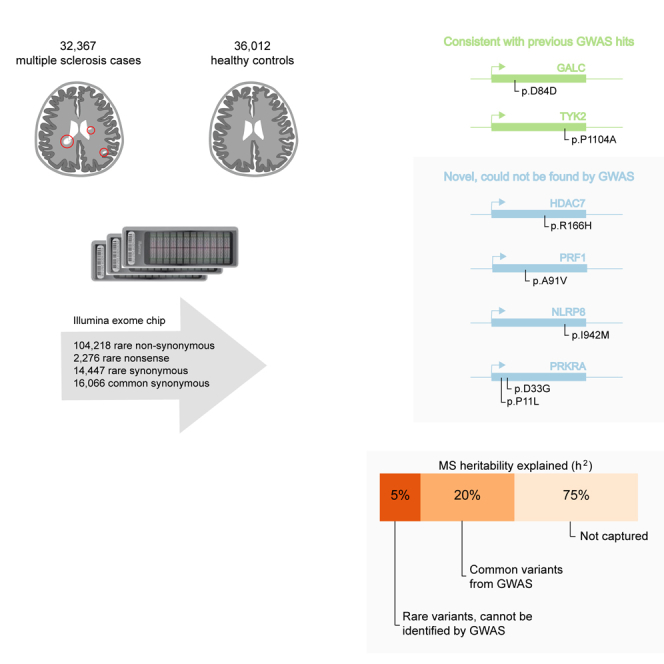
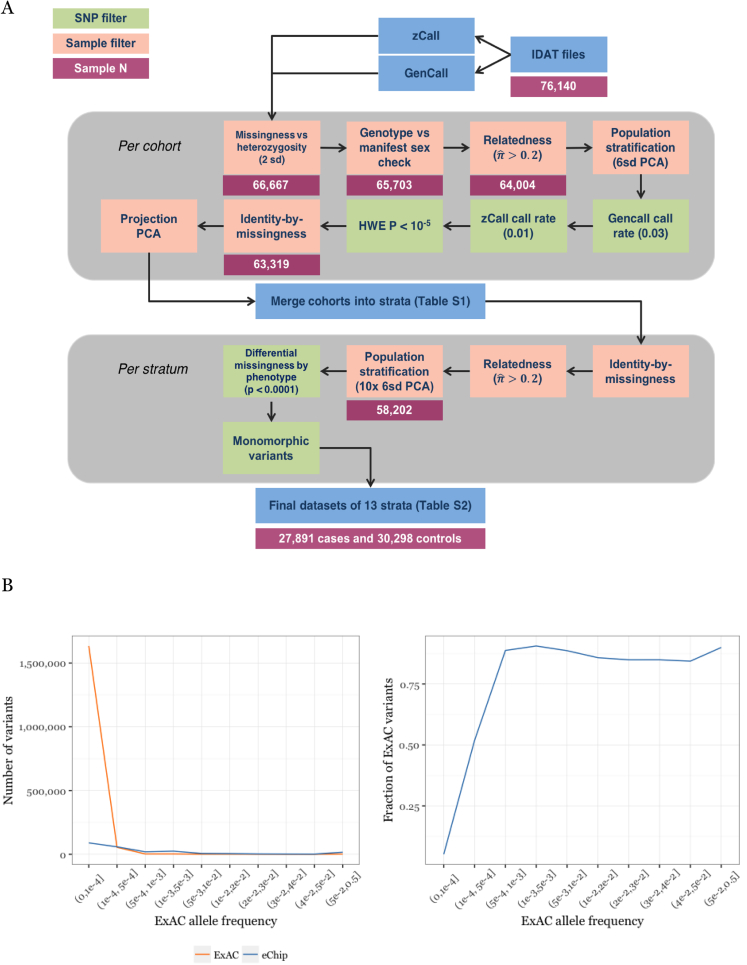
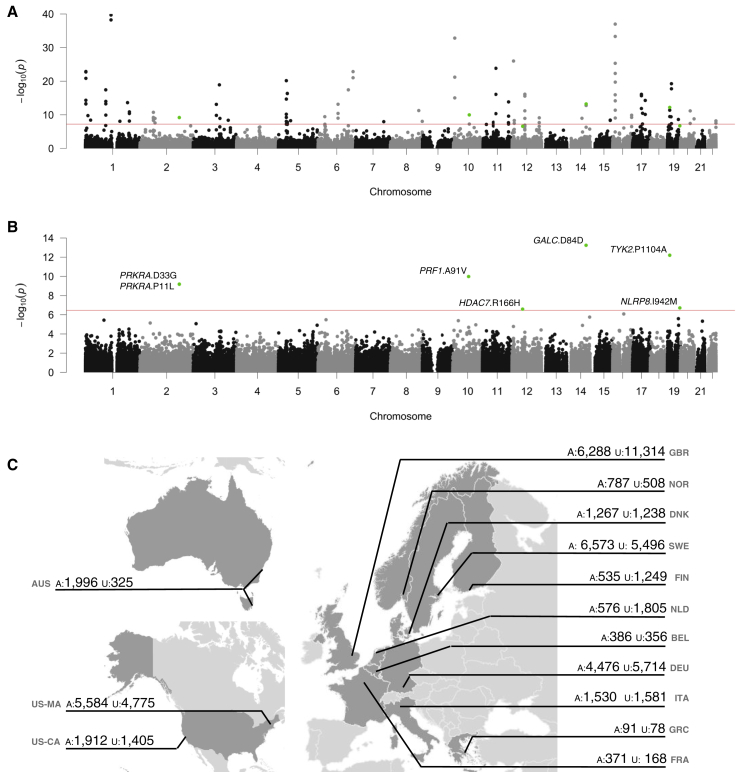
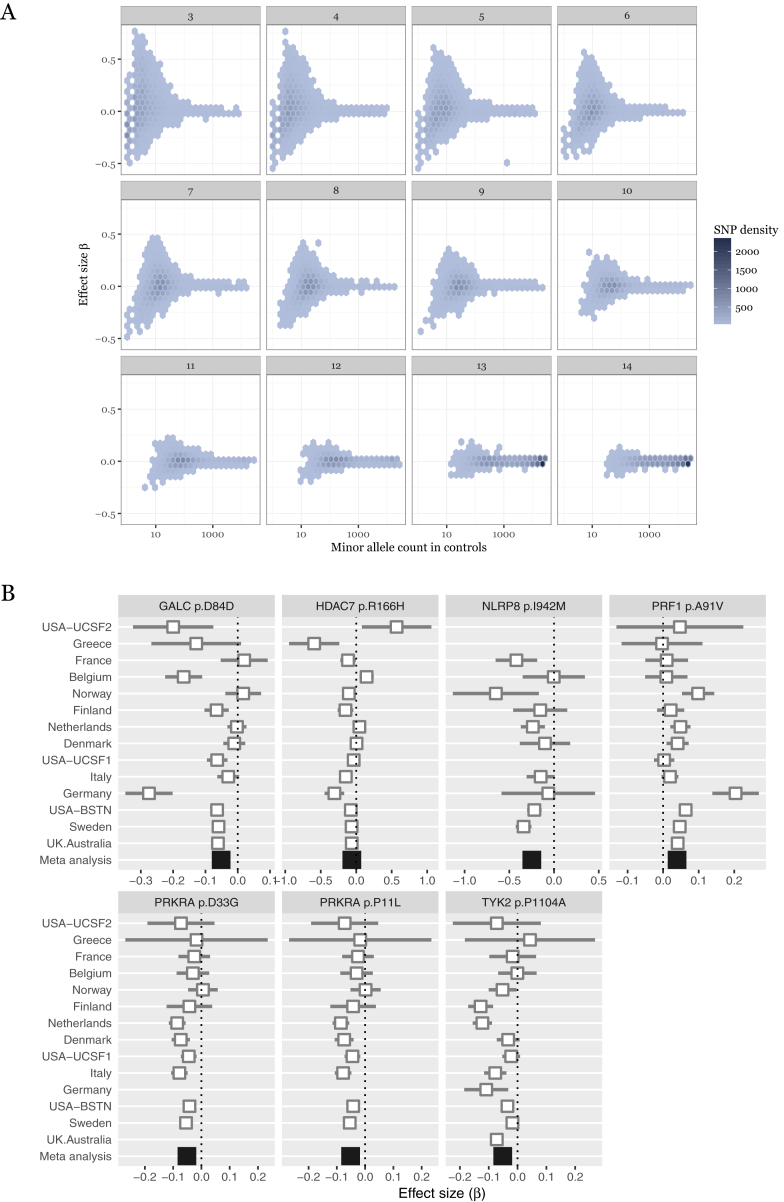
Multiple sclerosis is a complex neurological disease, with ∼20% of risk heritability attributable to common genetic variants, including >230 identified by genome-wide association studies. Multiple strands of evidence suggest that much of the remaining heritability is also due to additive effects of common variants rather than epistasis between these variants or mutations exclusive to individual families. Here, we show in 68,379 cases and controls that up to 5% of this heritability is explained by low-frequency variation in gene coding sequence. We identify four novel genes driving MS risk independently of common-variant signals, highlighting key pathogenic roles for regulatory T cell homeostasis and regulation, IFNγ biology, and NFκB signaling. As low-frequency variants do not show substantial linkage disequilibrium with other variants, and as coding variants are more interpretable and experimentally tractable than non-coding variation, our discoveries constitute a rich resource for dissecting the pathobiology of MS.
Copyright © 2018 The Author. Published by Elsevier Inc. All rights reserved.
Figures
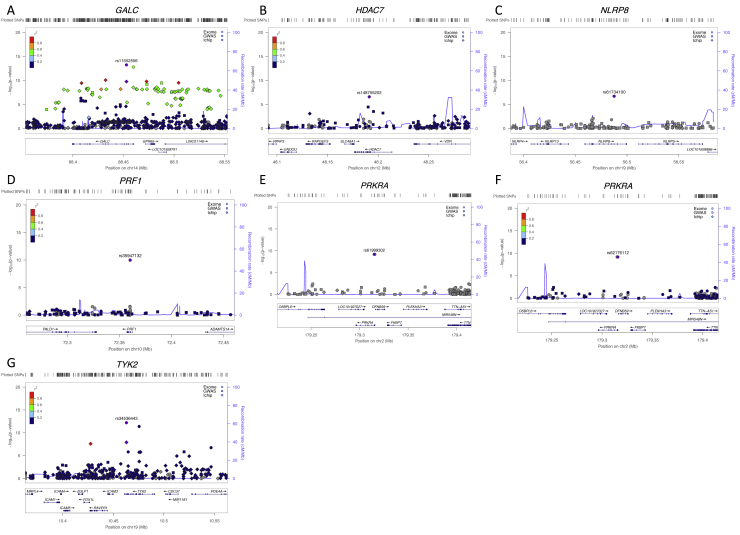
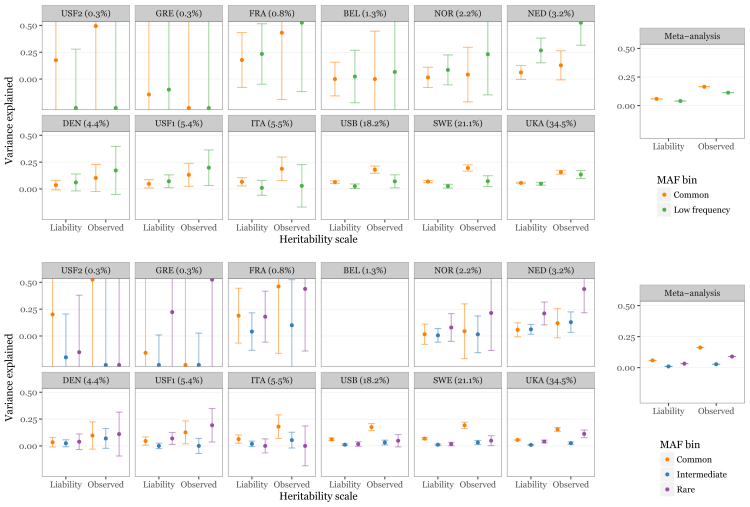
Comment in
-
New rare genetic variants in multiple sclerosis.J Neurol. 2019 Jan;266(1):278-280. doi: 10.1007/s00415-018-9128-9. J Neurol. 2019. PMID: 30460446 Free PMC article. No abstract available.
References
-
- Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene) Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat. Genet. 2009;41:824–828. - PubMed
Publication types
MeSH terms
Grants and funding
- R01 NS096212/NS/NINDS NIH HHS/United States
- G0000934/MRC_/Medical Research Council/United Kingdom
- R01 NS049477/NS/NINDS NIH HHS/United States
- 068545/Z/02/WT_/Wellcome Trust/United Kingdom
- DH_/Department of Health/United Kingdom
- 085475/B/08/Z/WT_/Wellcome Trust/United Kingdom
- G1100125/MRC_/Medical Research Council/United Kingdom
- U01 NS069208/NS/NINDS NIH HHS/United States
- R01 NS082347/NS/NINDS NIH HHS/United States
- 085475/Z/08/Z/WT_/Wellcome Trust/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- R01 NS026799/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
