

De novo assembly of haplotype-resolved genomes with trio binning
- PMID: 30346939
- PMCID: PMC6476705
- DOI: 10.1038/nbt.4277
De novo assembly of haplotype-resolved genomes with trio binning
Abstract
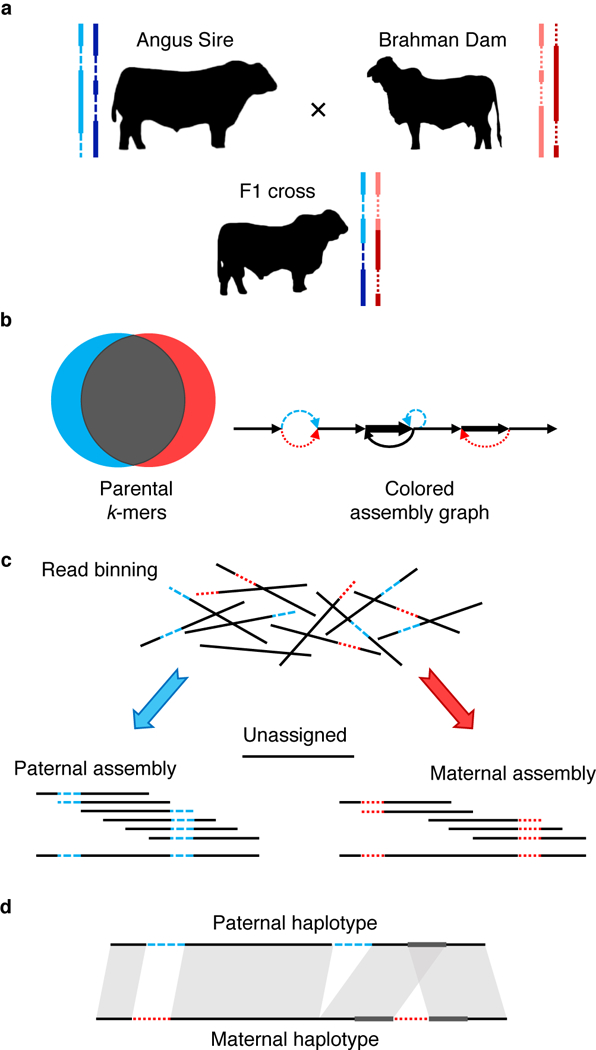
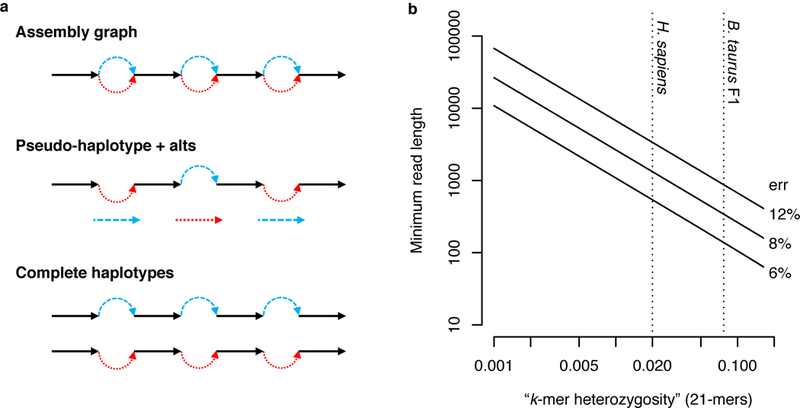
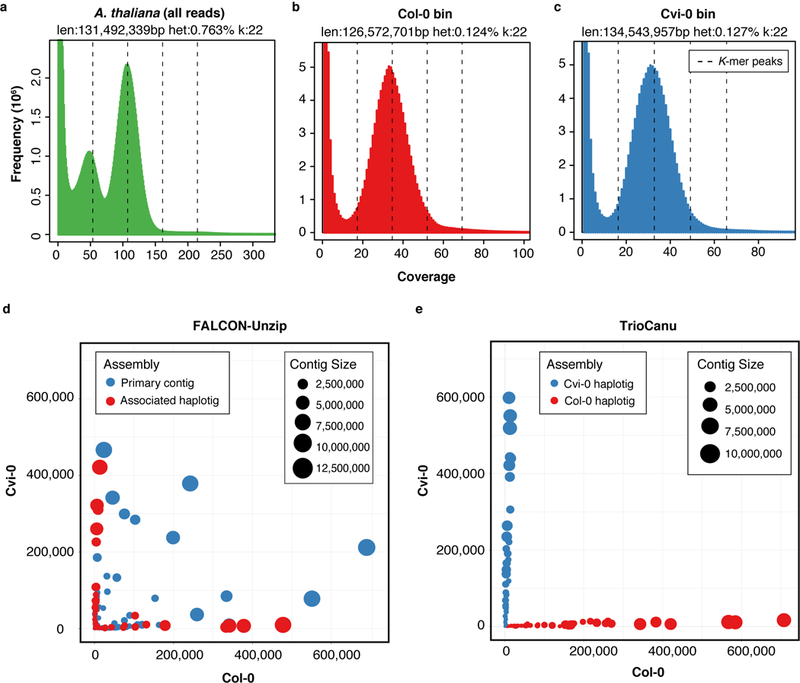
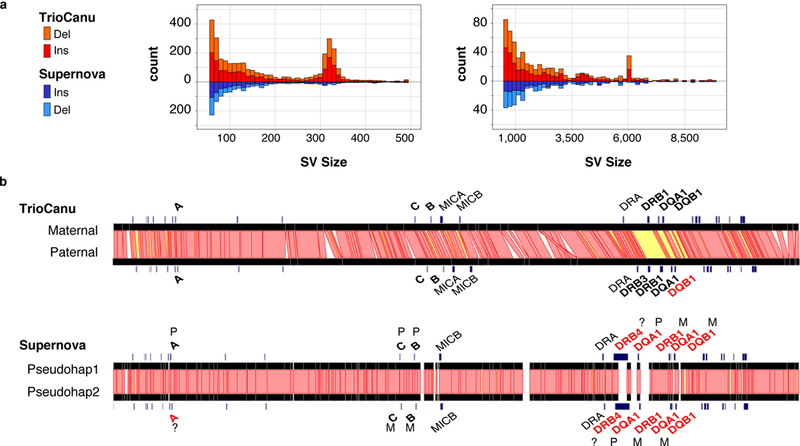
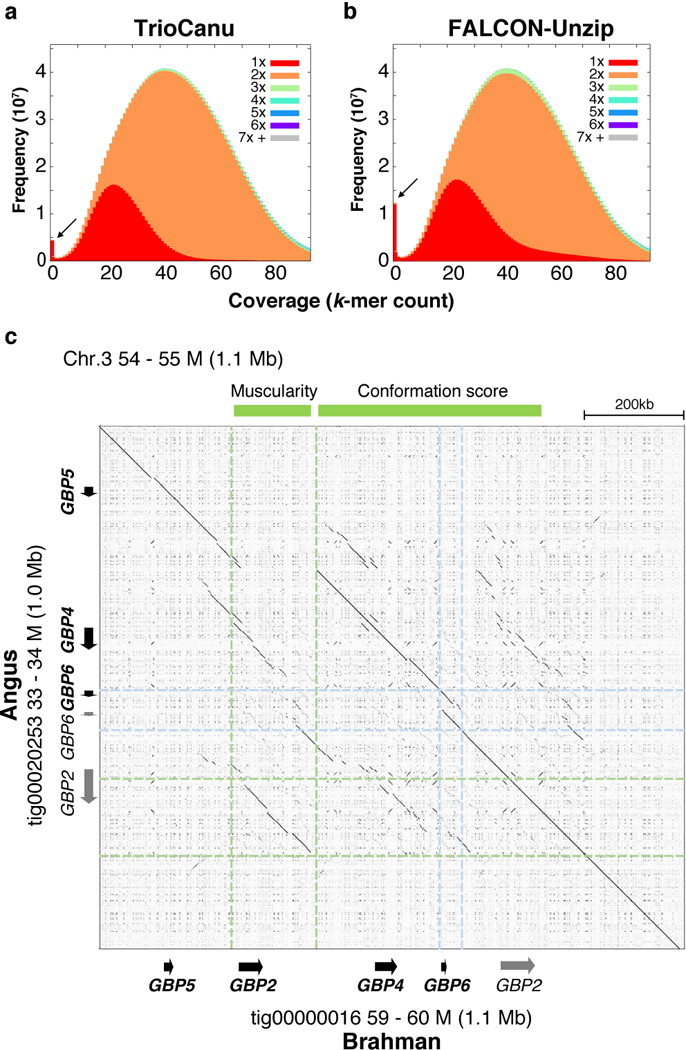
Complex allelic variation hampers the assembly of haplotype-resolved sequences from diploid genomes. We developed trio binning, an approach that simplifies haplotype assembly by resolving allelic variation before assembly. In contrast with prior approaches, the effectiveness of our method improved with increasing heterozygosity. Trio binning uses short reads from two parental genomes to first partition long reads from an offspring into haplotype-specific sets. Each haplotype is then assembled independently, resulting in a complete diploid reconstruction. We used trio binning to recover both haplotypes of a diploid human genome and identified complex structural variants missed by alternative approaches. We sequenced an F1 cross between the cattle subspecies Bos taurus taurus and Bos taurus indicus and completely assembled both parental haplotypes with NG50 haplotig sizes of >20 Mb and 99.998% accuracy, surpassing the quality of current cattle reference genomes. We suggest that trio binning improves diploid genome assembly and will facilitate new studies of haplotype variation and inheritance.
Conflict of interest statement
Competing financial interests
SBK is a current employee of Pacific Biosciences. All other authors declare no competing interests.
Figures
References
References for Online Methods
-
- Fofanov Y et al. How independent are the appearances of n-mers in different genomes? Bioinformatics 20, 2421–2428 (2004). - PubMed
-
- Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.399 7 (2013).
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
