

A k-mer-based method for the identification of phenotype-associated genomic biomarkers and predicting phenotypes of sequenced bacteria
- PMID: 30346947
- PMCID: PMC6211763
- DOI: 10.1371/journal.pcbi.1006434
A k-mer-based method for the identification of phenotype-associated genomic biomarkers and predicting phenotypes of sequenced bacteria
Abstract
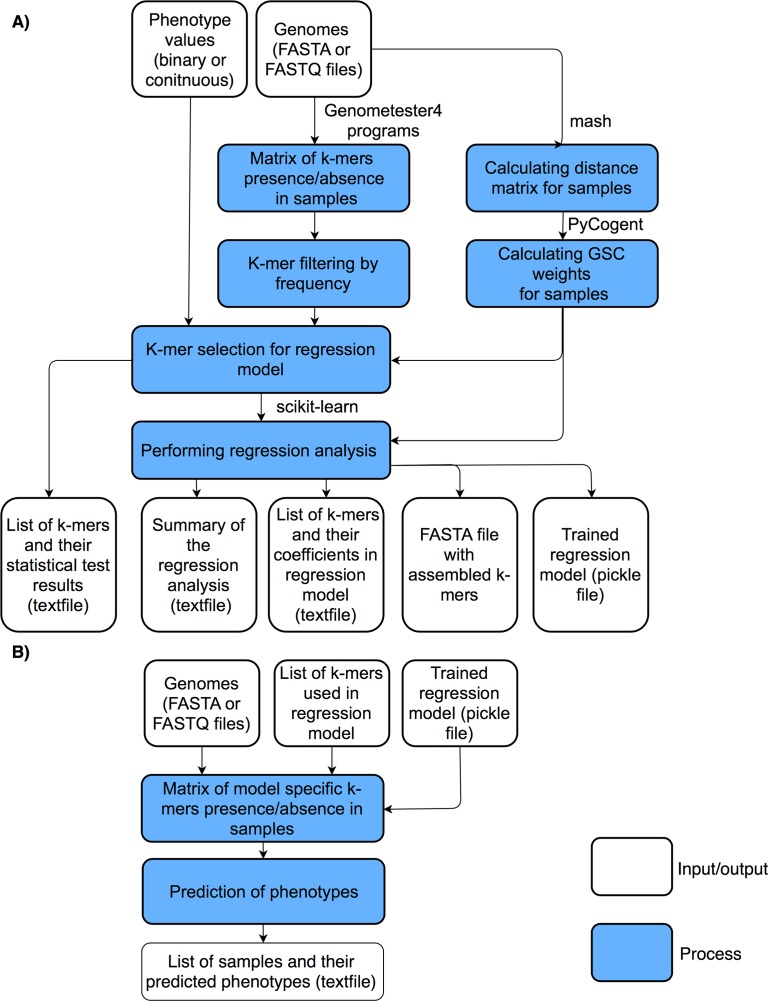
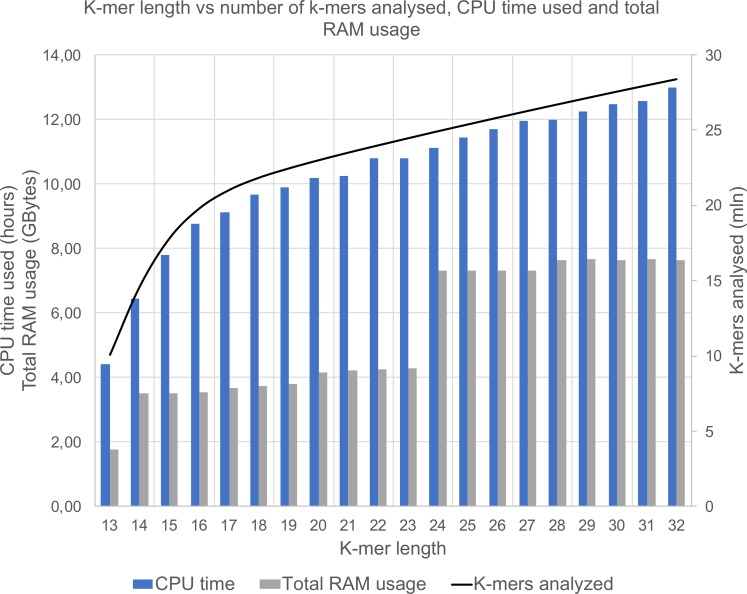
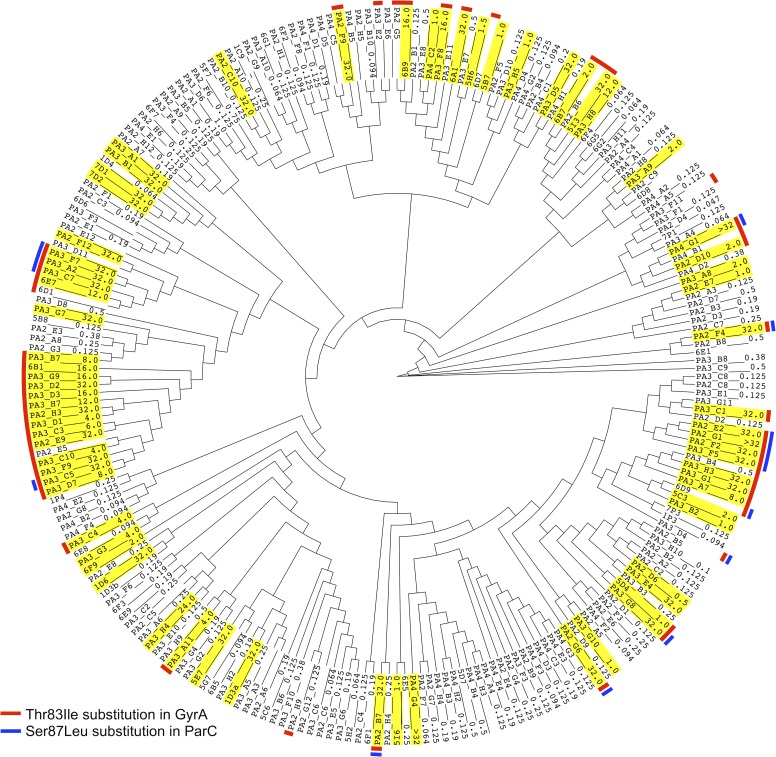
We have developed an easy-to-use and memory-efficient method called PhenotypeSeeker that (a) identifies phenotype-specific k-mers, (b) generates a k-mer-based statistical model for predicting a given phenotype and (c) predicts the phenotype from the sequencing data of a given bacterial isolate. The method was validated on 167 Klebsiella pneumoniae isolates (virulence), 200 Pseudomonas aeruginosa isolates (ciprofloxacin resistance) and 459 Clostridium difficile isolates (azithromycin resistance). The phenotype prediction models trained from these datasets obtained the F1-measure of 0.88 on the K. pneumoniae test set, 0.88 on the P. aeruginosa test set and 0.97 on the C. difficile test set. The F1-measures were the same for assembled sequences and raw sequencing data; however, building the model from assembled genomes is significantly faster. On these datasets, the model building on a mid-range Linux server takes approximately 3 to 5 hours per phenotype if assembled genomes are used and 10 hours per phenotype if raw sequencing data are used. The phenotype prediction from assembled genomes takes less than one second per isolate. Thus, PhenotypeSeeker should be well-suited for predicting phenotypes from large sequencing datasets. PhenotypeSeeker is implemented in Python programming language, is open-source software and is available at GitHub (https://github.com/bioinfo-ut/PhenotypeSeeker/).
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
-
- Bakour S, Sankar SA, Rathored J, Biagini P, Raoult D, Fournier P-E. Identification of virulence factors and antibiotic resistance markers using bacterial genomics. Future Microbiol [Internet]. 2016;11(3):455–66. Available from: http://www.futuremedicine.com/doi/10.2217/fmb.15.149 10.2217/fmb.15.149 - DOI - DOI - PubMed
-
- Wheeler NE, Gardner PP, Barquist L. Machine learning identifies signatures of host adaptation in the bacterial pathogen Salmonella enterica. PLOS Genet [Internet]. 2018;14(5):e1007333 Available from: http://dx.plos.org/10.1371/journal.pgen.1007333 10.1371/journal.pgen.1007333 - DOI - DOI - PMC - PubMed
-
- Li Y, Metcalf BJ, Chochua S, Li Z, Gertz RE, Walker H, et al. Validation of β-lactam minimum inhibitory concentration predictions for pneumococcal isolates with newly encountered penicillin binding protein (PBP) sequences. BMC Genomics. 2017;18(1):1–10. 10.1186/s12864-016-3406-7 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
