

Deleterious variation shapes the genomic landscape of introgression
- PMID: 30346959
- PMCID: PMC6233928
- DOI: 10.1371/journal.pgen.1007741
Deleterious variation shapes the genomic landscape of introgression
Abstract
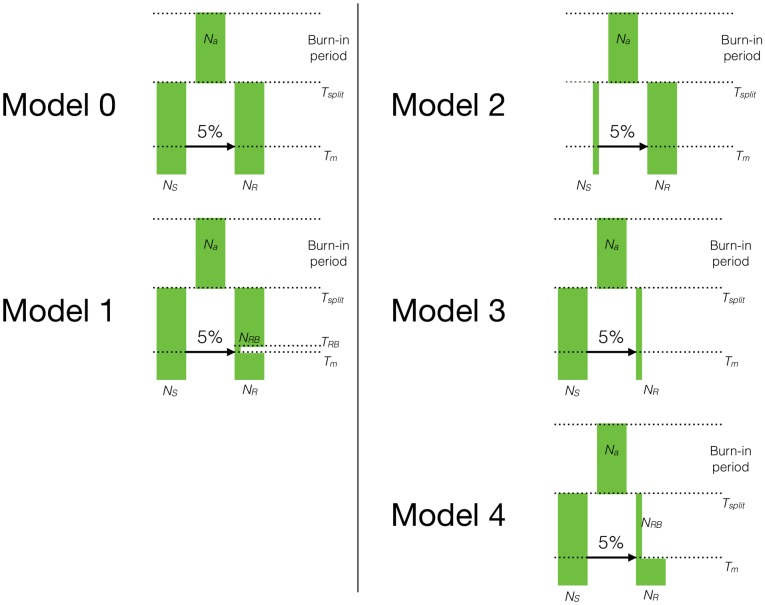
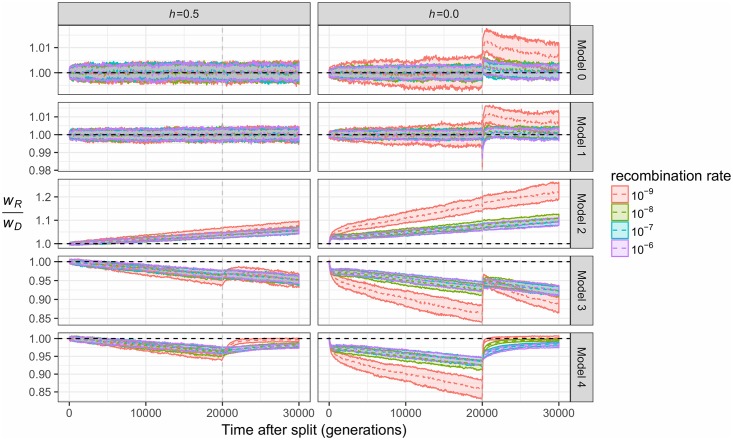
While it is appreciated that population size changes can impact patterns of deleterious variation in natural populations, less attention has been paid to how gene flow affects and is affected by the dynamics of deleterious variation. Here we use population genetic simulations to examine how gene flow impacts deleterious variation under a variety of demographic scenarios, mating systems, dominance coefficients, and recombination rates. Our results show that admixture between populations can temporarily reduce the genetic load of smaller populations and cause increases in the frequency of introgressed ancestry, especially if deleterious mutations are recessive. Additionally, when fitness effects of new mutations are recessive, between-population differences in the sites at which deleterious variants exist creates heterosis in hybrid individuals. Together, these factors lead to an increase in introgressed ancestry, particularly when recombination rates are low. Under certain scenarios, introgressed ancestry can increase from an initial frequency of 5% to 30-75% and fix at many loci, even in the absence of beneficial mutations. Further, deleterious variation and admixture can generate correlations between the frequency of introgressed ancestry and recombination rate or exon density, even in the absence of other types of selection. The direction of these correlations is determined by the specific demography and whether mutations are additive or recessive. Therefore, it is essential that null models of admixture include both demography and deleterious variation before invoking other mechanisms to explain unusual patterns of genetic variation.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures








Similar articles
-
Recombination Rate Variation, Hitchhiking, and Demographic History Shape Deleterious Load in Poplar.Mol Biol Evol. 2016 Nov;33(11):2899-2910. doi: 10.1093/molbev/msw169. Epub 2016 Aug 10. Mol Biol Evol. 2016. PMID: 27512114
-
The Impact of Recessive Deleterious Variation on Signals of Adaptive Introgression in Human Populations.Genetics. 2020 Jul;215(3):799-812. doi: 10.1534/genetics.120.303081. Epub 2020 Jun 2. Genetics. 2020. PMID: 32487519 Free PMC article.
-
Effects of Interference Between Selected Loci on the Mutation Load, Inbreeding Depression, and Heterosis.Genetics. 2015 Oct;201(2):745-57. doi: 10.1534/genetics.115.178533. Epub 2015 Aug 12. Genetics. 2015. PMID: 26269503 Free PMC article.
-
Genomic inference using diffusion models and the allele frequency spectrum.Curr Opin Genet Dev. 2018 Dec;53:140-147. doi: 10.1016/j.gde.2018.10.001. Epub 2018 Oct 23. Curr Opin Genet Dev. 2018. PMID: 30366252 Review.
-
Population Genomic Scans for Natural Selection and Demography.Annu Rev Genet. 2024 Nov;58(1):319-339. doi: 10.1146/annurev-genet-111523-102651. Epub 2024 Nov 14. Annu Rev Genet. 2024. PMID: 39227130 Review.
Cited by
-
Associative Overdominance and Negative Epistasis Shape Genome-Wide Ancestry Landscape in Supplemented Fish Populations.Genes (Basel). 2021 Apr 3;12(4):524. doi: 10.3390/genes12040524. Genes (Basel). 2021. PMID: 33916757 Free PMC article.
-
Repeated Mitochondrial Capture With Limited Genomic Introgression in a Lizard Group.Mol Ecol. 2025 May;34(10):e17766. doi: 10.1111/mec.17766. Epub 2025 Apr 16. Mol Ecol. 2025. PMID: 40241380 Free PMC article.
-
The temporal and genomic scale of selection following hybridization.bioRxiv [Preprint]. 2023 May 26:2023.05.25.542345. doi: 10.1101/2023.05.25.542345. bioRxiv. 2023. Update in: Proc Natl Acad Sci U S A. 2024 Mar 19;121(12):e2309168121. doi: 10.1073/pnas.2309168121. PMID: 37337589 Free PMC article. Updated. Preprint.
-
Adaptive Introgression as an Evolutionary Force: A Meta-Analysis of Knowledge Trends.Evol Appl. 2025 Jun 20;18(6):e70103. doi: 10.1111/eva.70103. eCollection 2025 Jun. Evol Appl. 2025. PMID: 40548230 Free PMC article.
-
Constraining models of dominance for nonsynonymous mutations in the human genome.PLoS Genet. 2024 Sep 20;20(9):e1011198. doi: 10.1371/journal.pgen.1011198. eCollection 2024 Sep. PLoS Genet. 2024. PMID: 39302992 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
