

A Tumor-Promoting Phorbol Ester Causes a Large Increase in APOBEC3A Expression and a Moderate Increase in APOBEC3B Expression in a Normal Human Keratinocyte Cell Line without Increasing Genomic Uracils
- PMID: 30348839
- PMCID: PMC6290372
- DOI: 10.1128/MCB.00238-18
A Tumor-Promoting Phorbol Ester Causes a Large Increase in APOBEC3A Expression and a Moderate Increase in APOBEC3B Expression in a Normal Human Keratinocyte Cell Line without Increasing Genomic Uracils
Abstract
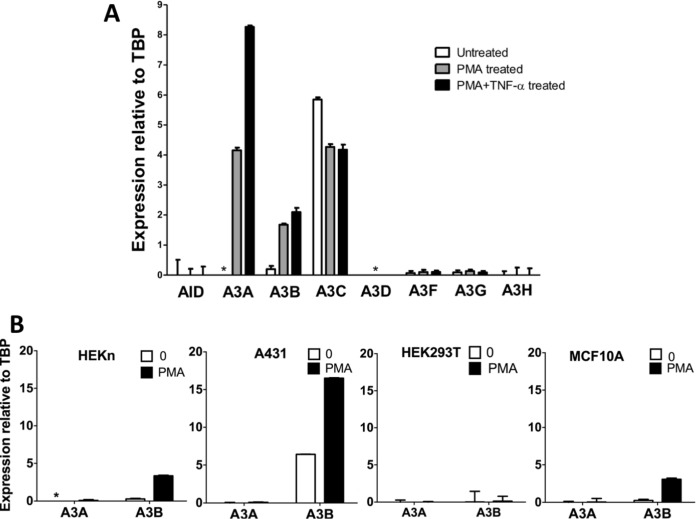
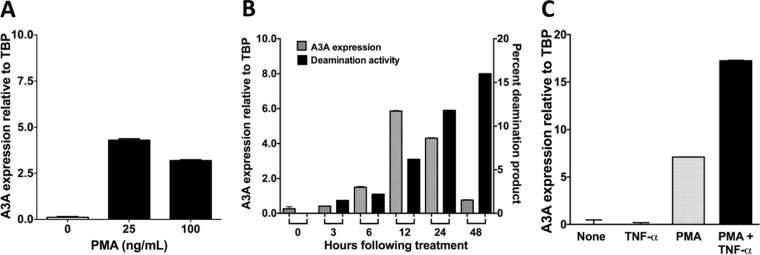
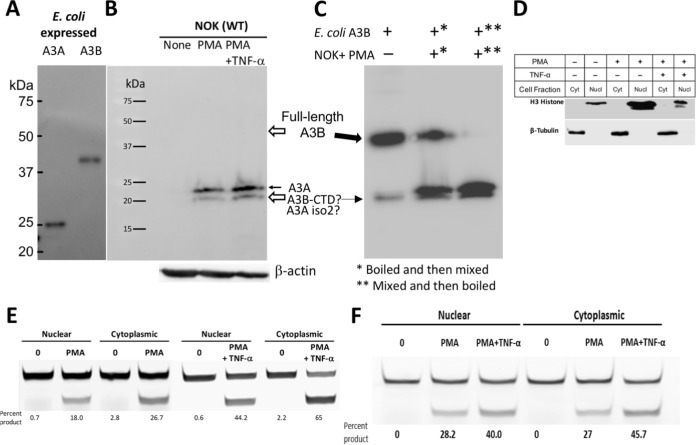
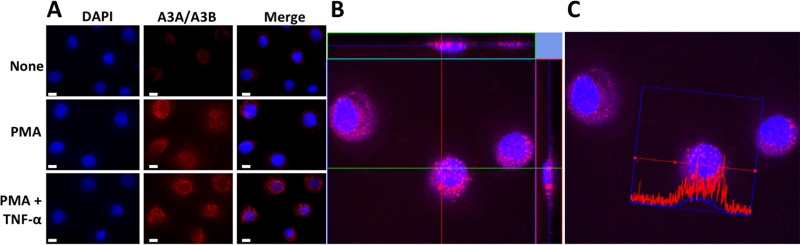
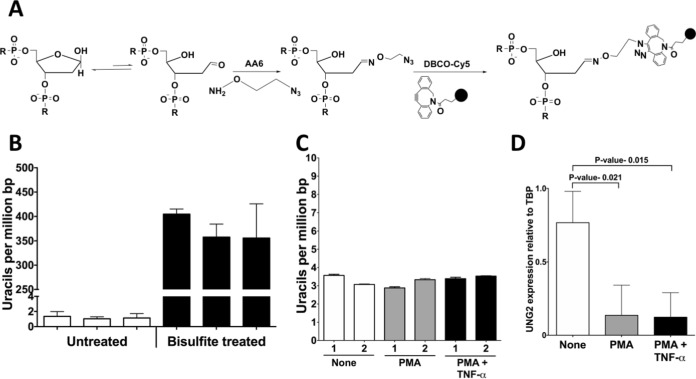
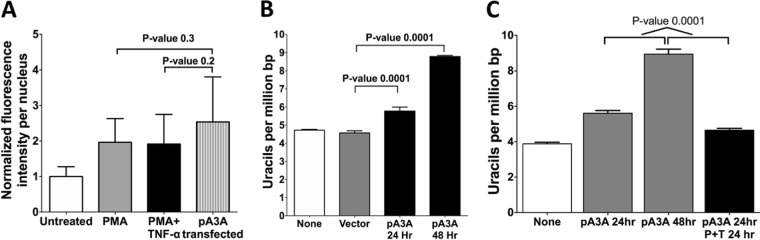
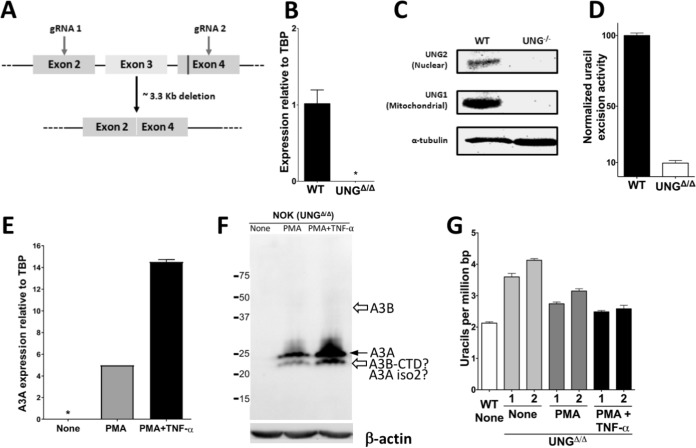
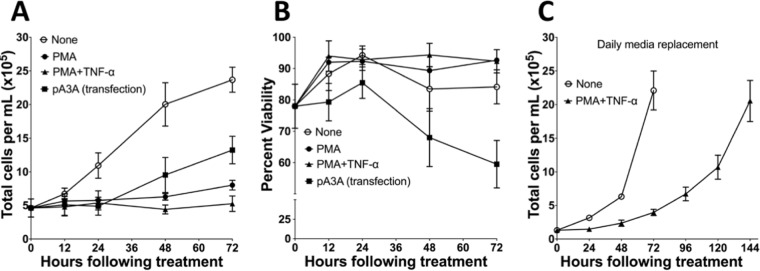
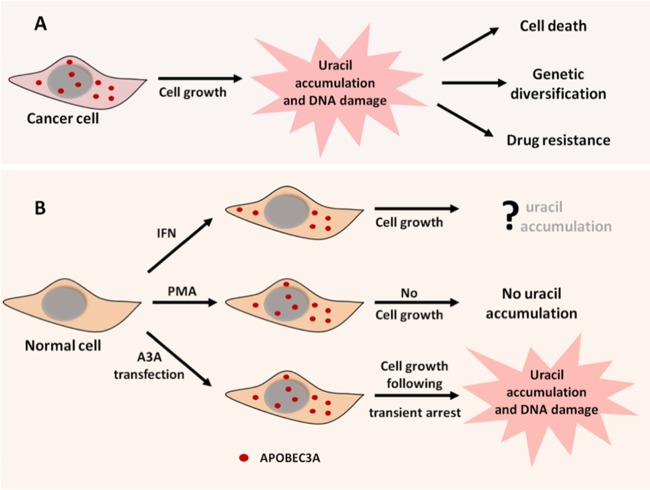
Phorbol 12-myristate 13-acetate (PMA) promotes skin cancer in rodents. The mutations found in murine tumors are similar to those found in human skin cancers, and PMA promotes proliferation of human skin cells. PMA treatment of human keratinocytes increases the synthesis of APOBEC3A, an enzyme that converts cytosines in single-stranded DNA to uracil, and mutations in a variety of human cancers are attributed to APOBEC3A or APOBEC3B expression. We tested here the possibility that induction of APOBEC3A by PMA causes genomic accumulation of uracils that may lead to such mutations. When a human keratinocyte cell line was treated with PMA, both APOBEC3A and APOBEC3B gene expression increased, anti-APOBEC3A/APOBEC3B antibody bound a protein(s) in the nucleus, and nuclear extracts displayed cytosine deamination activity. Surprisingly, there was little increase in genomic uracils in PMA-treated wild-type or uracil repair-defective cells. In contrast, cells transfected with a plasmid expressing APOBEC3A acquired more genomic uracils. Unexpectedly, PMA treatment, but not APOBEC3A plasmid transfection, caused a cessation in cell growth. Hence, a reduction in single-stranded DNA at replication forks may explain the inability of PMA-induced APOBEC3A/APOBEC3B to increase genomic uracils. These results suggest that the proinflammatory PMA is unlikely to promote extensive APOBEC3A/APOBEC3B-mediated cytosine deaminations in human keratinocytes.
Keywords: APOBEC3A; phorbol myristate acetate; psoriasis; replication fork and DNA damage; uracils in DNA.
Copyright © 2018 American Society for Microbiology.
Figures
Similar articles
-
Visualization of uracils created by APOBEC3A using UdgX shows colocalization with RPA at stalled replication forks.Nucleic Acids Res. 2020 Nov 18;48(20):e118. doi: 10.1093/nar/gkaa845. Nucleic Acids Res. 2020. PMID: 33074285 Free PMC article.
-
Distinguishing preferences of human APOBEC3A and APOBEC3B for cytosines in hairpin loops, and reflection of these preferences in APOBEC-signature cancer genome mutations.Nat Commun. 2024 Mar 18;15(1):2369. doi: 10.1038/s41467-024-46231-w. Nat Commun. 2024. PMID: 38499553 Free PMC article.
-
Mesoscale DNA features impact APOBEC3A and APOBEC3B deaminase activity and shape tumor mutational landscapes.Nat Commun. 2024 Mar 18;15(1):2370. doi: 10.1038/s41467-024-45909-5. Nat Commun. 2024. PMID: 38499542 Free PMC article.
-
APOBEC3B: pathological consequences of an innate immune DNA mutator.Biomed J. 2015 Mar-Apr;38(2):102-10. doi: 10.4103/2319-4170.148904. Biomed J. 2015. PMID: 25566802 Review.
-
Roles of APOBEC3A and APOBEC3B in Human Papillomavirus Infection and Disease Progression.Viruses. 2017 Aug 21;9(8):233. doi: 10.3390/v9080233. Viruses. 2017. PMID: 28825669 Free PMC article. Review.
Cited by
-
Activation of DNA damage repair factors in HPV positive oropharyngeal cancers.Virology. 2020 Aug;547:27-34. doi: 10.1016/j.virol.2020.05.003. Epub 2020 May 22. Virology. 2020. PMID: 32560902 Free PMC article.
-
Genome-wide mapping of regions preferentially targeted by the human DNA-cytosine deaminase APOBEC3A using uracil-DNA pulldown and sequencing.J Biol Chem. 2019 Oct 11;294(41):15037-15051. doi: 10.1074/jbc.RA119.008053. Epub 2019 Aug 19. J Biol Chem. 2019. PMID: 31431505 Free PMC article.
-
Human activation-induced deaminase lacks strong replicative strand bias or preference for cytosines in hairpin loops.Nucleic Acids Res. 2022 May 20;50(9):5145-5157. doi: 10.1093/nar/gkac296. Nucleic Acids Res. 2022. PMID: 35524550 Free PMC article.
-
Detection of Genomic Uracil Patterns.Int J Mol Sci. 2021 Apr 9;22(8):3902. doi: 10.3390/ijms22083902. Int J Mol Sci. 2021. PMID: 33918885 Free PMC article. Review.
-
Aberrant APOBEC3B Expression in Breast Cancer Is Linked to Proliferation and Cell Cycle Phase.Cells. 2023 Apr 18;12(8):1185. doi: 10.3390/cells12081185. Cells. 2023. PMID: 37190094 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources