

Chromatin run-on and sequencing maps the transcriptional regulatory landscape of glioblastoma multiforme
- PMID: 30349114
- PMCID: PMC6204104
- DOI: 10.1038/s41588-018-0244-3
Chromatin run-on and sequencing maps the transcriptional regulatory landscape of glioblastoma multiforme
Abstract
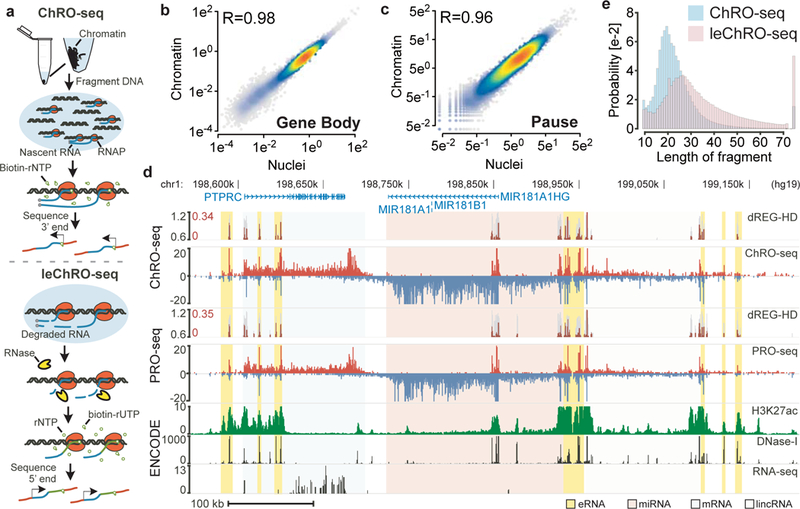
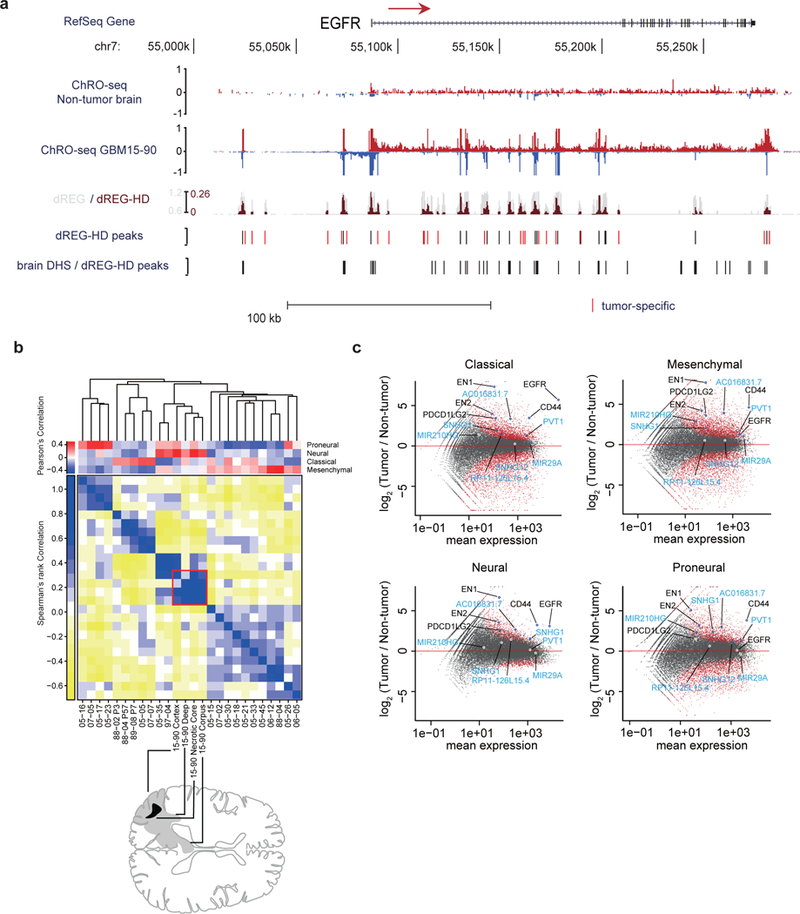
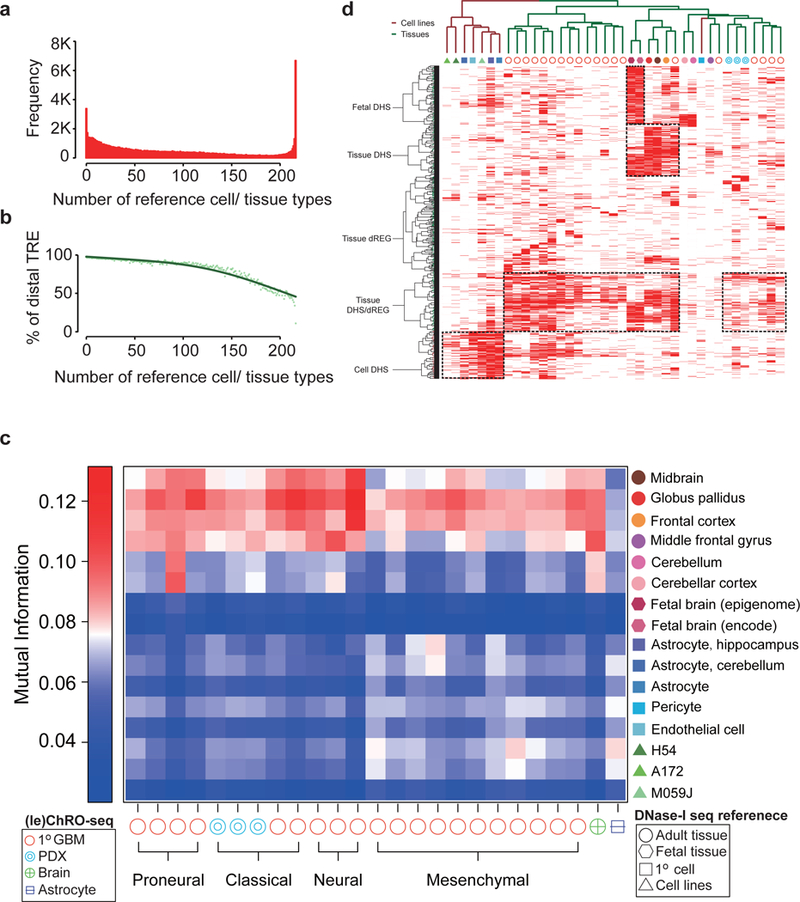
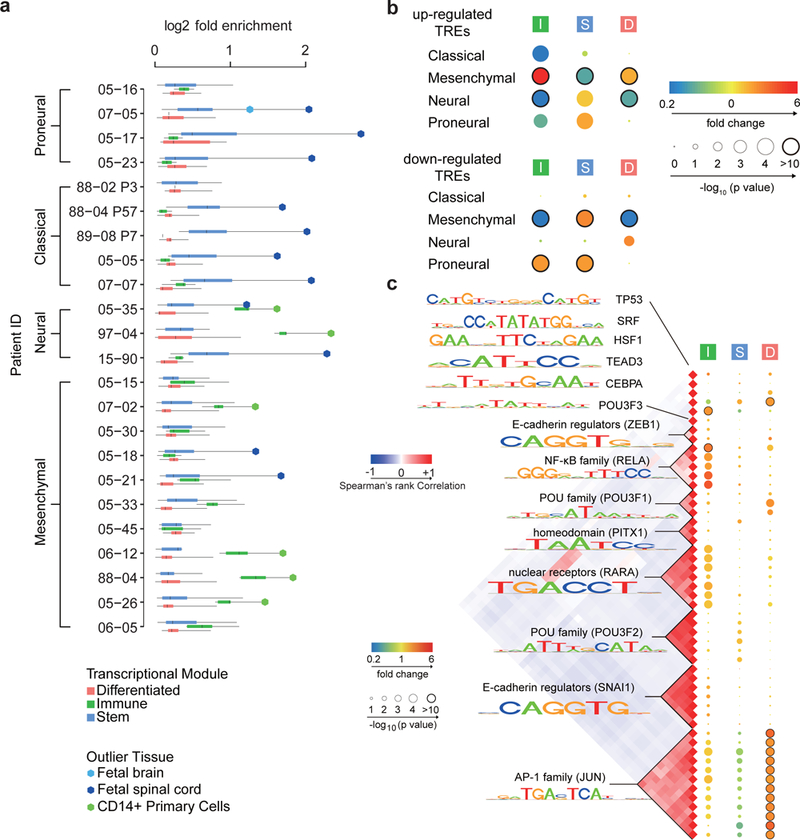
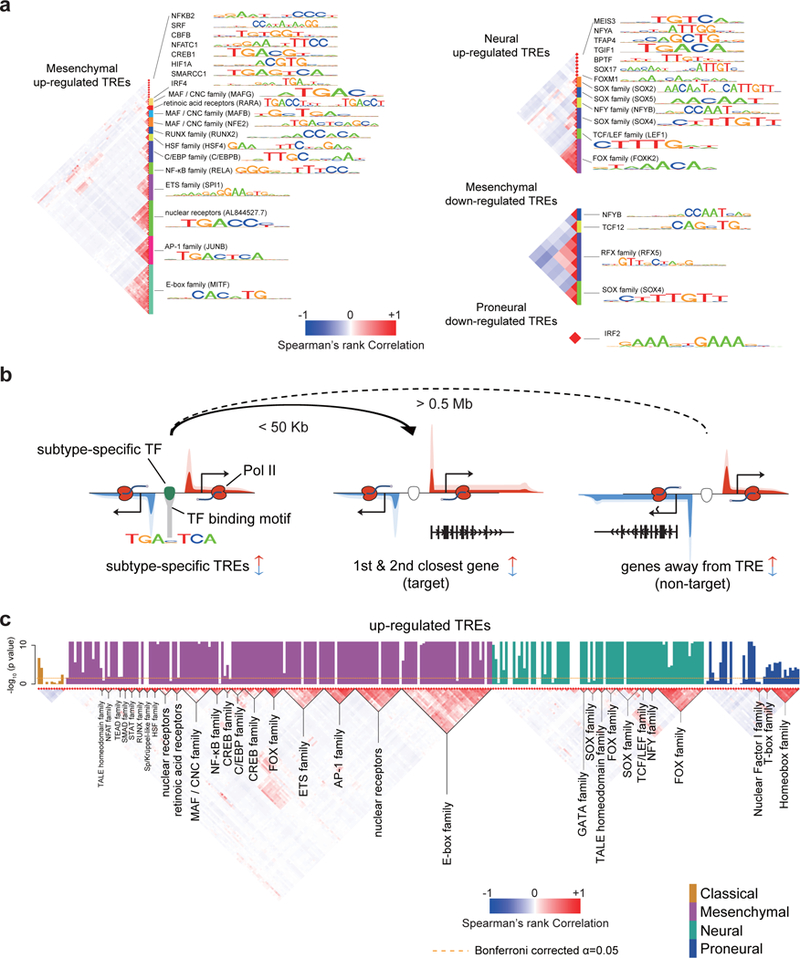
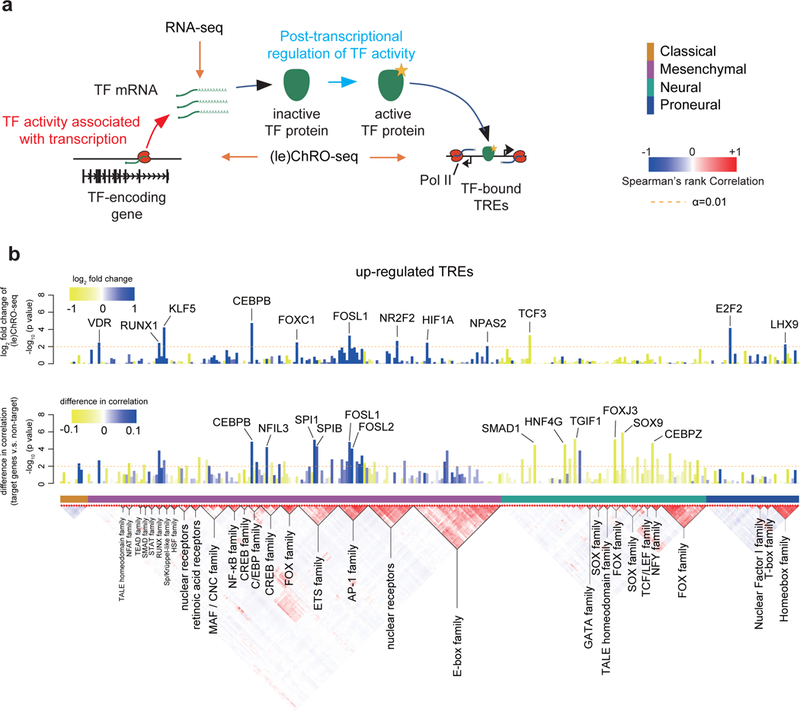
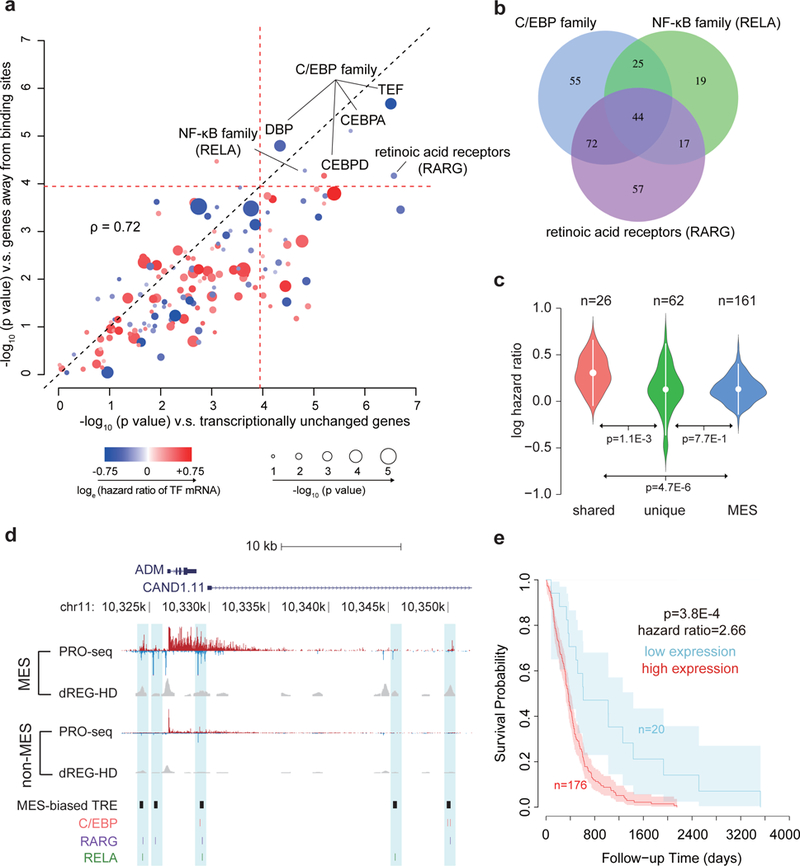
The human genome encodes a variety of poorly understood RNA species that remain challenging to identify using existing genomic tools. We developed chromatin run-on and sequencing (ChRO-seq) to map the location of RNA polymerase for almost any input sample, including samples with degraded RNA that are intractable to RNA sequencing. We used ChRO-seq to map nascent transcription in primary human glioblastoma (GBM) brain tumors. Enhancers identified in primary GBMs resemble open chromatin in the normal human brain. Rare enhancers that are activated in malignant tissue drive regulatory programs similar to the developing nervous system. We identified enhancers that regulate groups of genes that are characteristic of each known GBM subtype and transcription factors that drive them. Finally we discovered a core group of transcription factors that control the expression of genes associated with clinical outcomes. This study characterizes the transcriptional landscape of GBM and introduces ChRO-seq as a method to map regulatory programs that contribute to complex diseases.
Conflict of interest statement
Competing financial interests:
The authors declare no competing financial interests.
Figures
References
-
- Cheng J et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308, 1149–1154 (2005). - PubMed
References (Online Methods):
-
- Canute GW et al. Hydroxyurea accelerates the loss of epidermal growth factor receptor genes amplified as double-minute chromosomes in human glioblastoma multiforme. Neurosurgery 39, 976–983 (1996). - PubMed
-
- Eller JL, Longo SL, Hicklin DJ & Canute GW Activity of anti-epidermal growth factor receptor monoclonal antibody C225 against glioblastoma multiforme. Neurosurgery 51, 1005–13; discussion 1013–4 (2002). - PubMed
-
- Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
