

Computational analysis and prediction of lysine malonylation sites by exploiting informative features in an integrative machine-learning framework
- PMID: 30351377
- PMCID: PMC6954445
- DOI: 10.1093/bib/bby079
Computational analysis and prediction of lysine malonylation sites by exploiting informative features in an integrative machine-learning framework
Abstract
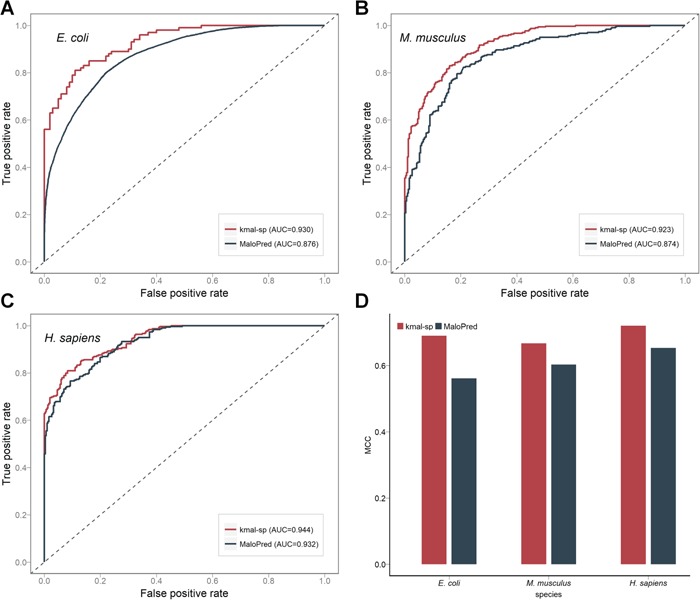
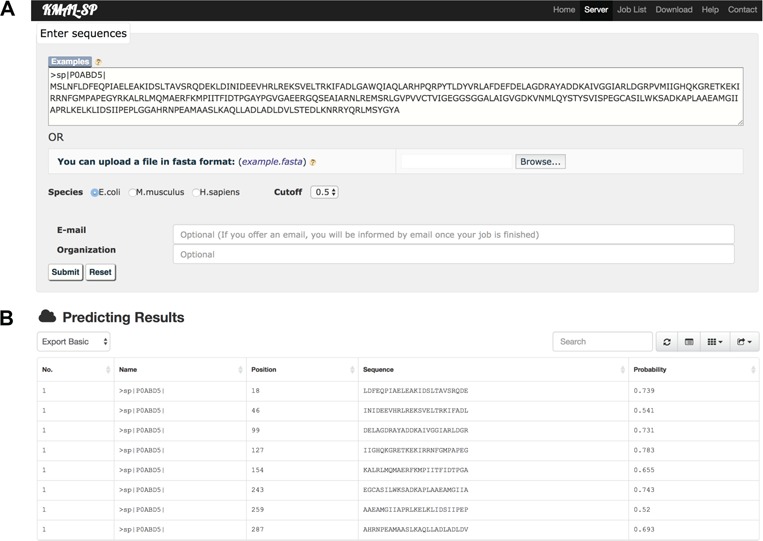
As a newly discovered post-translational modification (PTM), lysine malonylation (Kmal) regulates a myriad of cellular processes from prokaryotes to eukaryotes and has important implications in human diseases. Despite its functional significance, computational methods to accurately identify malonylation sites are still lacking and urgently needed. In particular, there is currently no comprehensive analysis and assessment of different features and machine learning (ML) methods that are required for constructing the necessary prediction models. Here, we review, analyze and compare 11 different feature encoding methods, with the goal of extracting key patterns and characteristics from residue sequences of Kmal sites. We identify optimized feature sets, with which four commonly used ML methods (random forest, support vector machines, K-nearest neighbor and logistic regression) and one recently proposed [Light Gradient Boosting Machine (LightGBM)] are trained on data from three species, namely, Escherichia coli, Mus musculus and Homo sapiens, and compared using randomized 10-fold cross-validation tests. We show that integration of the single method-based models through ensemble learning further improves the prediction performance and model robustness on the independent test. When compared to the existing state-of-the-art predictor, MaloPred, the optimal ensemble models were more accurate for all three species (AUC: 0.930, 0.923 and 0.944 for E. coli, M. musculus and H. sapiens, respectively). Using the ensemble models, we developed an accessible online predictor, kmal-sp, available at http://kmalsp.erc.monash.edu/. We hope that this comprehensive survey and the proposed strategy for building more accurate models can serve as a useful guide for inspiring future developments of computational methods for PTM site prediction, expedite the discovery of new malonylation and other PTM types and facilitate hypothesis-driven experimental validation of novel malonylated substrates and malonylation sites.
Keywords: Light Gradient Boosting Machine; computational prediction; ensemble learning; feature encoding methods; lysine malonylation; machine learning.
© The Author(s) 2018. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
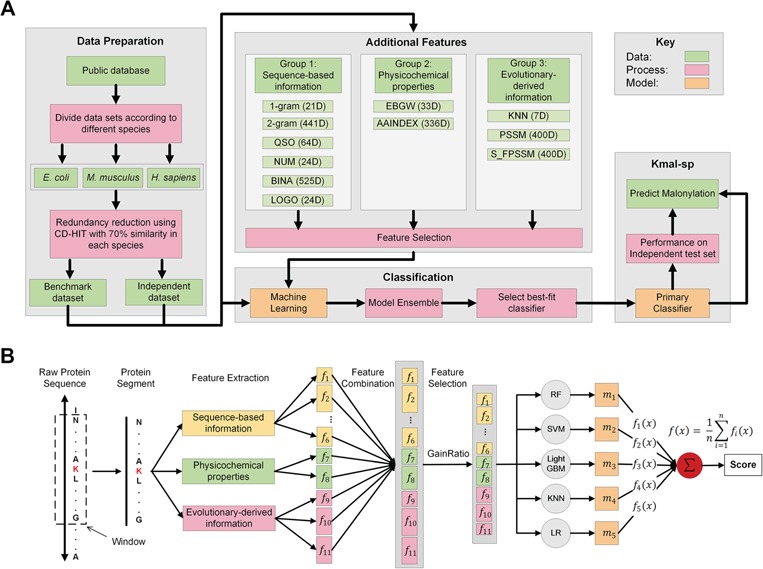
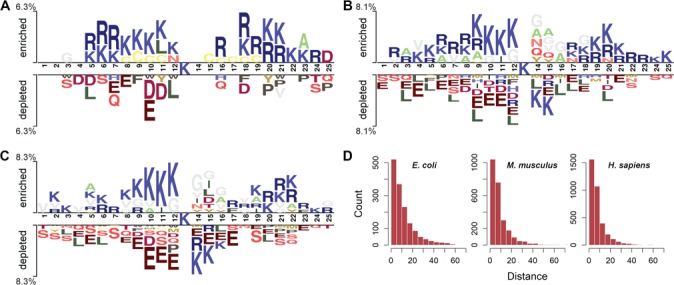
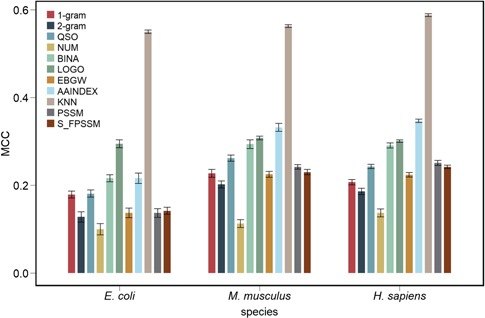
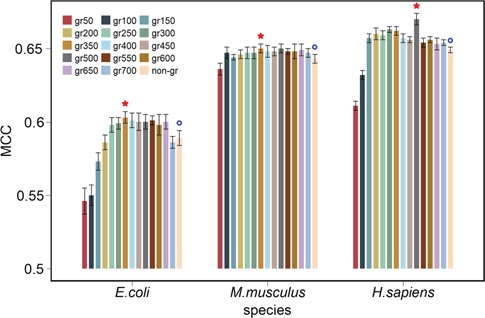
References
-
- Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol 2007;8:139–48. - PubMed
-
- Westermann S, Weber K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol 2003;4:938–47. - PubMed
-
- Harmel R, Fiedler D. Features and regulation of non-enzymatic post-translational modifications. Nat Chem Biol 2018;14:244–52. - PubMed
-
- Johnson LN. The regulation of protein phosphorylation. Biochem Soc Trans 2009;37:627–41. - PubMed
-
- Ambler RP, Rees MW. Epsilon-N-Methyl-lysine in bacterial flagellar protein. Nature 1959;183:1654–5. - PubMed
