

Germline SDHB and SDHD mutations in pheochromocytoma and paraganglioma patients
- PMID: 30352407
- PMCID: PMC6240141
- DOI: 10.1530/EC-18-0325
Germline SDHB and SDHD mutations in pheochromocytoma and paraganglioma patients
Abstract
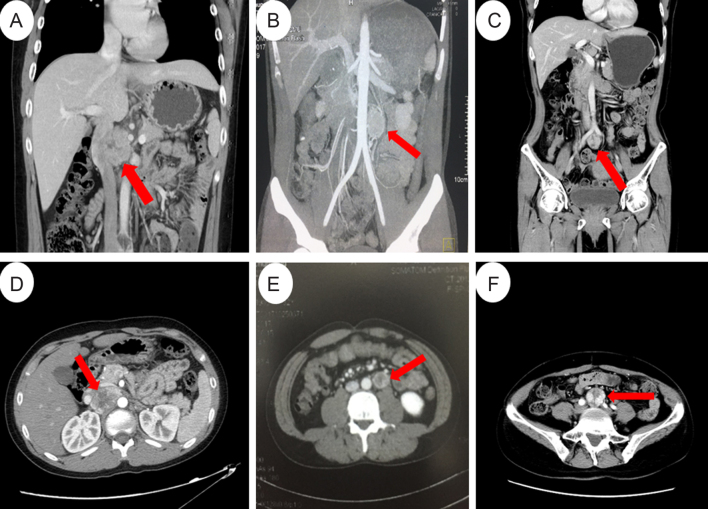
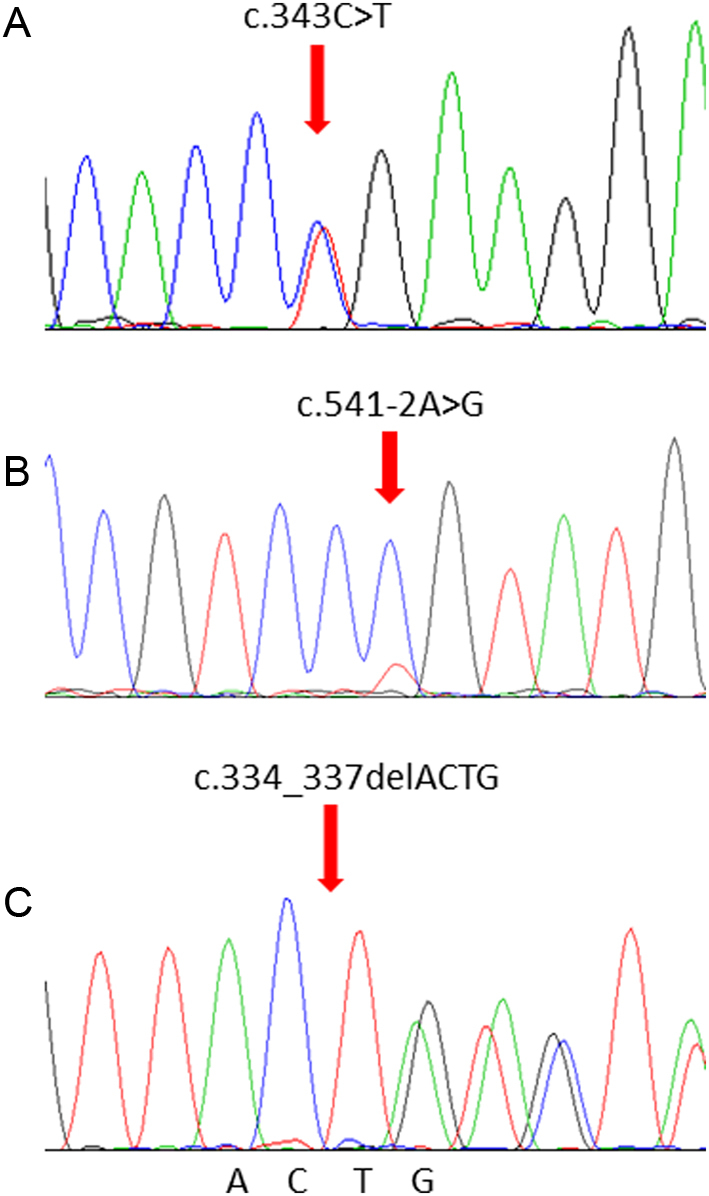
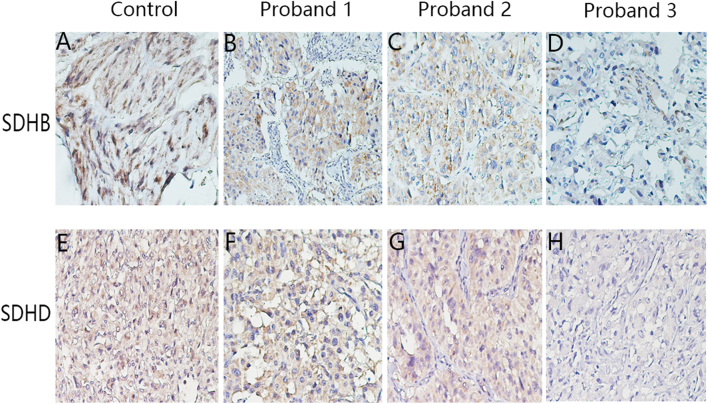
Pheochromocytoma and paragangliomas (PCC/PGL) are neuroendocrine tumors that arise from chromaffin cells of the adrenal medulla and sympathetic/parasympathetic ganglia, respectively. Of clinical relevance regarding diagnosis is the highly variable presentation of symptoms in PCC/PGL patients. To date, the clear-cut correlations between the genotypes and phenotypes of PCC/PGL have not been entirely established. In this study, we reviewed the medical records of PCC/PGL patients with pertinent clinical, laboratory and genetic information. Next-generation sequencing (NGS) performed on patient samples revealed specific germline mutations in the SDHB (succinate dehydrogenase complex iron-sulfur subunit B) and SDHD (succinate dehydrogenase complex subunit D) genes and these mutations were validated by Sanger sequencing. Of the 119 patients, two were identified with SDHB mutation and one with SDHD mutation. Immunohistochemical (IHC) staining was used to analyze the expression of these mutated genes. The germline mutations identified in the SDH genes were c343C>T and c.541-542A>G in the SDHB gene and c.334-337delACTG in the SDHD gene. IHC staining of tumors from the c.343C>T and c.541-2A>G carriers showed positive expression of SDHB. Tumors from the c.334-337delACTG carrier showed no expression of SDHD and a weak diffused staining pattern for SDHB. We strongly recommend genetic testing for suspected PCC/PGL patients with a positive family history, early onset of age, erratic hypertension, recurrence or multiple tumor sites and loss of SDHB and/or SDHD expression. Tailored personal management should be conducted once a patient is confirmed as an SDHB and/or SDHD mutation carrier or diagnosed with PCC/PGL.
Keywords: PCC/PGL; SDHB; SDHD; genotype–phenotype correlation.
Figures
References
-
- Jochmanova I, Wolf KI, King KS, Nambuba J, Wesley R, Martucci V, Raygada M, Adams KT, Prodanov T, Fojo AT, et al SDHB-related pheochromocytoma and paraganglioma penetrance and genotype-phenotype correlations. Journal of Cancer Research and Clinical Oncology 2017. 143 1421–1435. ( 10.1007/s00432-017-2397-3) - DOI - PMC - PubMed
-
- Korpershoek E, van Nederveen FH, Komminoth P, de Krijger RR. Familial endocrine tumours: pheochromocytomas and extra-adrenal paragangliomas – an update. Diagnostic Histopathology 2017. 23 335–345. ( 10.1016/j.mpdhp.2017.06.001) - DOI
-
- Benn D, Gimenez-Roqueplo A, Reilly J, Bertherat J, Burgess J, Byth K, Croxson M, Dahia P, Elston M, Gimm O, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. Journal of Clinical Endocrinology and Metabolism 2006. 91 827–836. ( 10.1210/jc.2005-1862) - DOI - PubMed
-
- Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clinic Proceedings 1983. 58 802–804. - PubMed
LinkOut - more resources
Full Text Sources
