

A new hepatitis E virus genotype 2 strain identified from an outbreak in Nigeria, 2017
- PMID: 30352598
- PMCID: PMC6199738
- DOI: 10.1186/s12985-018-1082-8
A new hepatitis E virus genotype 2 strain identified from an outbreak in Nigeria, 2017
Abstract
Background: In 2017 the Nigerian Ministry of Health notified the World Health Organization (WHO) of an outbreak of hepatitis E located in the north-east region of the country with 146 cases with 2 deaths. The analysis of the hepatitis E virus (HEV) genotypes responsible for the outbreak revealed the predominance of HEV genotypes 1 (HEV-1) and 2 (HEV-2). Molecular data of HEV-2 genomes are limited; therefore we characterized a HEV-2 strain of the outbreak in more detail.
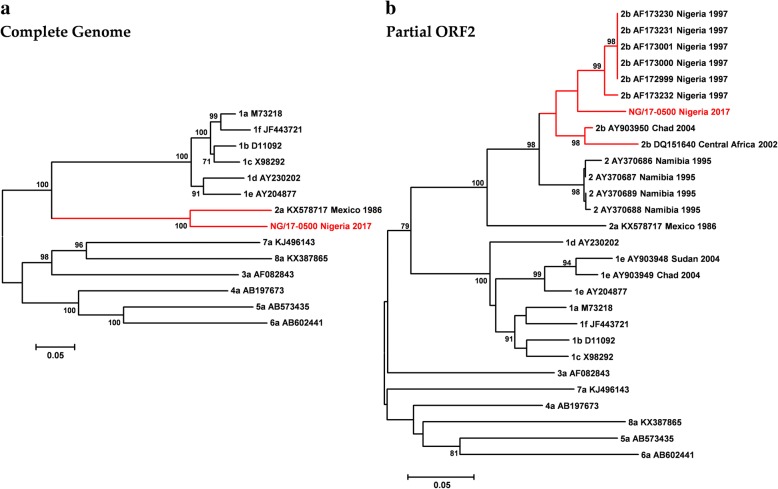
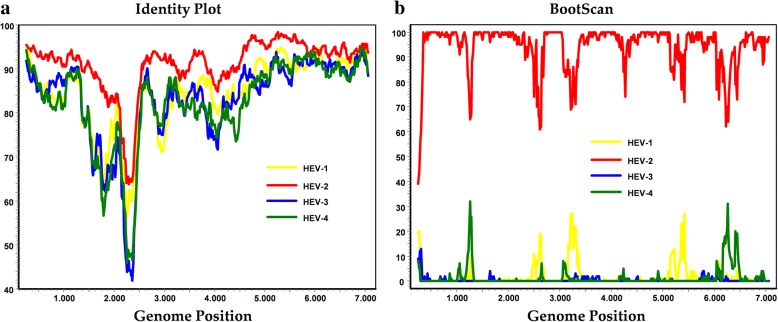
Finding: The full-length genome sequence of an HEV-2 strain (NG/17-0500) from the outbreak was amplified using newly designed consensus primers. Comparison with other HEV complete genome sequences, including the only HEV-2 strain (Mex-14) with available complete genome sequences and the availability of data of partial HEV-2 sequences from Sub-Saharan Africa, suggests that NG/17-0500 belongs to HEV subtype 2b (HEV-2b).
Conclusions: We identified a novel HEV-2b strain from Sub-Saharan Africa, which is the second complete HEV-2 sequence to date, whose natural history and epidemiology merit further investigation.
Keywords: Complete genome; HEV genotype 2; HEV subtype 2b; Hepatitis E virus; Nigeria; Outbreak.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable. German public health institute (RKI) is authorized to receive blood residuals from diagnostics for surveillance purposes (Infection Protection Act IfSG §13). All samples analysed were anonymised.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Lee GH, Tan BH, Teo EC, Lim SG, Dan YY, Wee A, Aw PP, Zhu Y, Hibberd ML, Tan CK, et al. Chronic infection with camelid hepatitis E virus in a liver transplant recipient who regularly consumes camel meat and milk. Gastroenterology. 2016;150(2):355–357. doi: 10.1053/j.gastro.2015.10.048. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- Partnership in Postgraduate Education/Global Health Protection, RKI (GE)/International
- GOARN Program/WHO_/World Health Organization/International
- Schloarship/China Scholarship Council/International
- 001/WHO_/World Health Organization/International
- 91611817/Deutscher Akademischer Austauschdienst/International
LinkOut - more resources
Full Text Sources
