

Activation of KRAS Mediates Resistance to Targeted Therapy in MET Exon 14-mutant Non-small Cell Lung Cancer
- PMID: 30352902
- PMCID: PMC6377821
- DOI: 10.1158/1078-0432.CCR-18-1640
Activation of KRAS Mediates Resistance to Targeted Therapy in MET Exon 14-mutant Non-small Cell Lung Cancer
Abstract
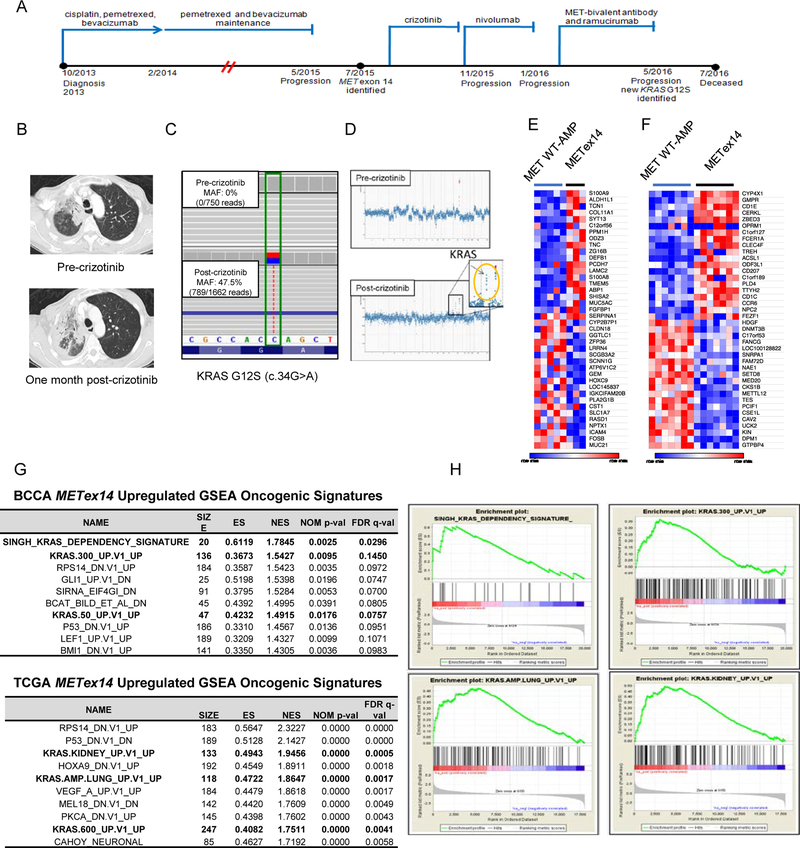
Purpose: MET exon 14 splice site alterations that cause exon skipping at the mRNA level (METex14) are actionable oncogenic drivers amenable to therapy with MET tyrosine kinase inhibitors (TKI); however, secondary resistance eventually arises in most cases while other tumors display primary resistance. Beyond relatively uncommon on-target MET kinase domain mutations, mechanisms underlying primary and acquired resistance remain unclear.
Experimental design: We examined clinical and genomic data from 113 patients with lung cancer with METex14. MET TKI resistance due to KRAS mutation was functionally evaluated using in vivo and in vitro models.
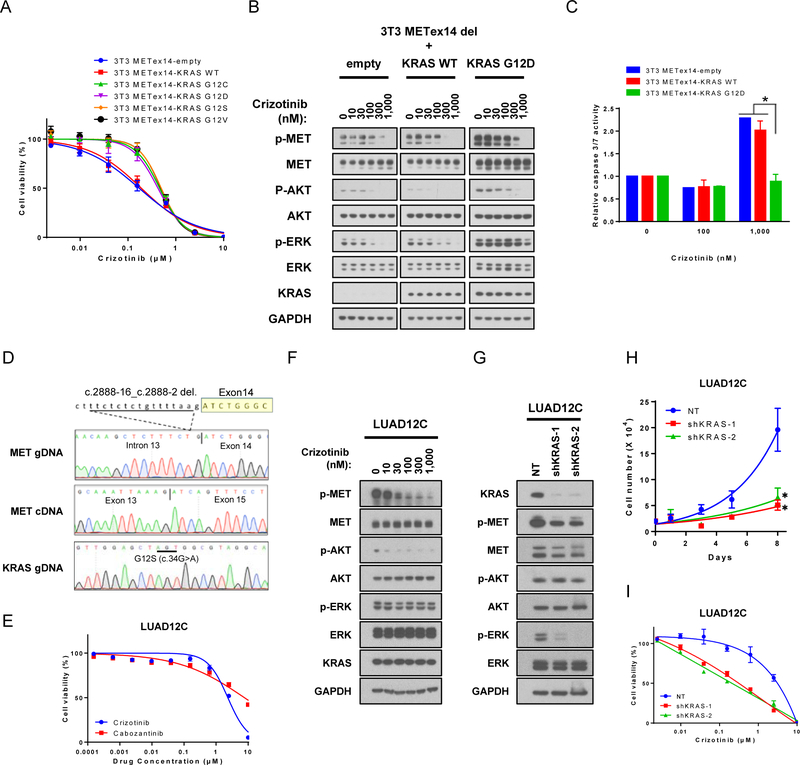
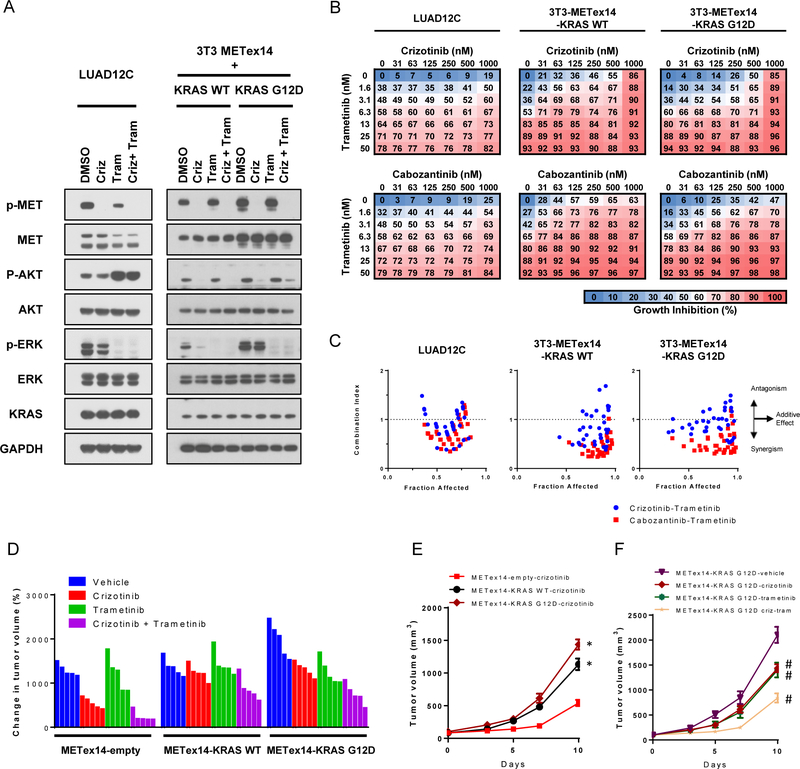
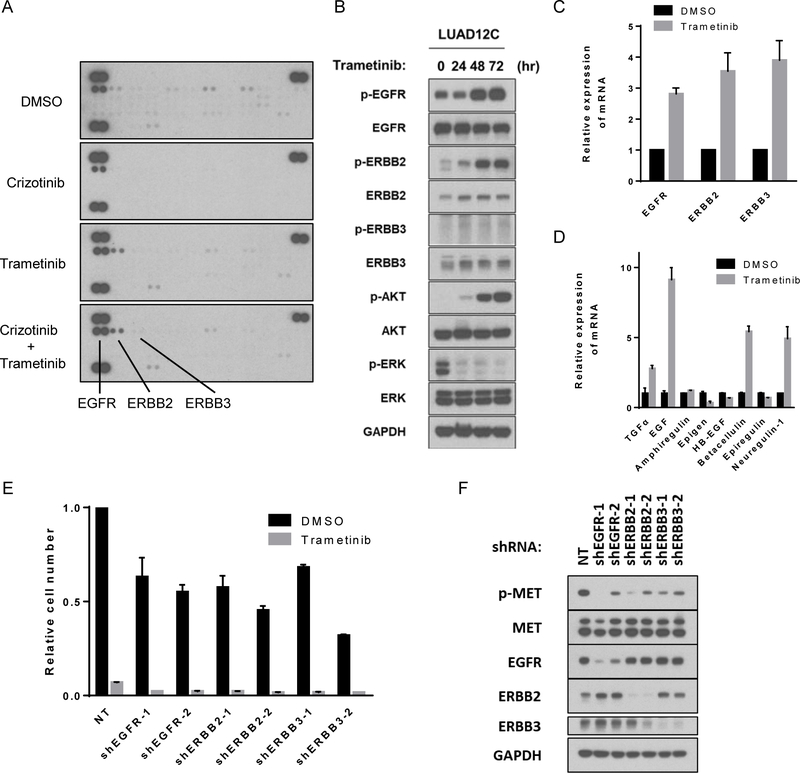
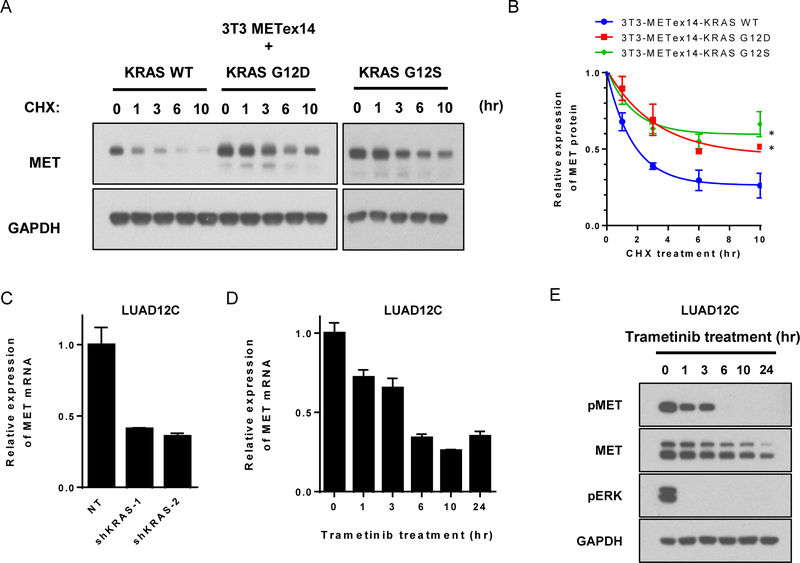
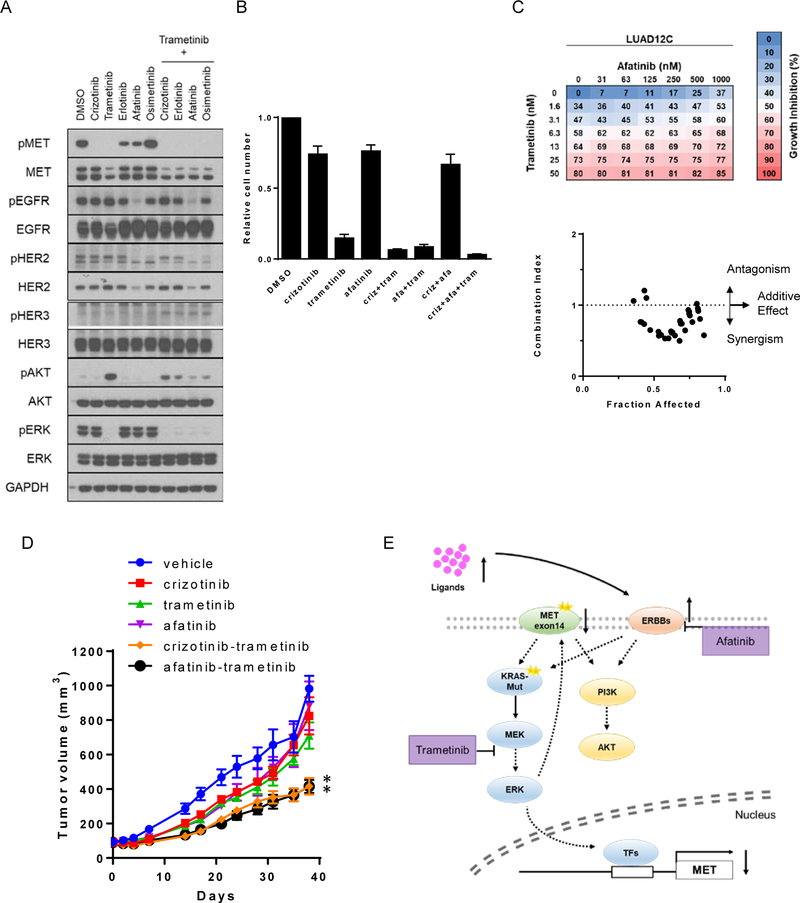
Results: Five of 113 patients (4.4%) with METex14 had concurrent KRAS G12 mutations, a rate of KRAS cooccurrence significantly higher than in other major driver-defined lung cancer subsets. In one patient, the KRAS mutation was acquired post-crizotinib, while the remaining 4 METex14 patients harbored the KRAS mutation prior to MET TKI therapy. Gene set enrichment analysis of transcriptomic data from lung cancers with METex14 revealed preferential activation of the KRAS pathway. Moreover, expression of oncogenic KRAS enhanced MET expression. Using isogenic and patient-derived models, we show that KRAS mutation results in constitutive activation of RAS/ERK signaling and resistance to MET inhibition. Dual inhibition of MET or EGFR/ERBB2 and MEK reduced growth of cell line and xenograft models.
Conclusions: KRAS mutation is a recurrent mechanism of primary and secondary resistance to MET TKIs in METex14 lung cancers. Dual inhibition of MET or EGFR/ERBB2 and MEK may represent a potential therapeutic approach in this molecular cohort.
©2018 American Association for Cancer Research.
Conflict of interest statement
Conflict of interest:
Charles M. Rudin is a consultant for Bristol-Myers Squibb, Abbvie, Seattle Genetics, Harpoon Therapeutics, Genentech Roche, and AstraZeneca.
Mark G. Kris is a consultant for Ariad, AstraZeneca and Genentech Roche and received research funding from Genentech Roche and Puma Biotechnology.
Alexander Drilon is a consultant for Ignyta, LOXO Oncology, TP Therapeutics, AstraZeneca, Pfizer, Blueprint Medicines, Genentech Roche, Takeda, and has received research funding from Foundation Medicine.
Marc Ladanyi has received advisory board compensation from Boehringer Ingelheim, AstraZeneca, Bristol-Myers Squibb, Takeda, and Bayer, and research support from LOXO Oncology and Helsinn Healthcare.
Romel Somwar has received research support from Helsinn Healthcare
All other authors do not report any relevant conflicts of interest
Figures
References
-
- Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–48. - PubMed
-
- Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103. - PubMed
-
- Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat Med. 2016;22:1314–20. - PubMed
-
- Ma PC, Kijima T, Maulik G, Fox EA, Sattler M, Griffin JD, et al. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63:6272–81. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
