

Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease
- PMID: 30355078
- PMCID: PMC6207260
- DOI: 10.1161/CIRCRESAHA.118.312563
Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease
Abstract
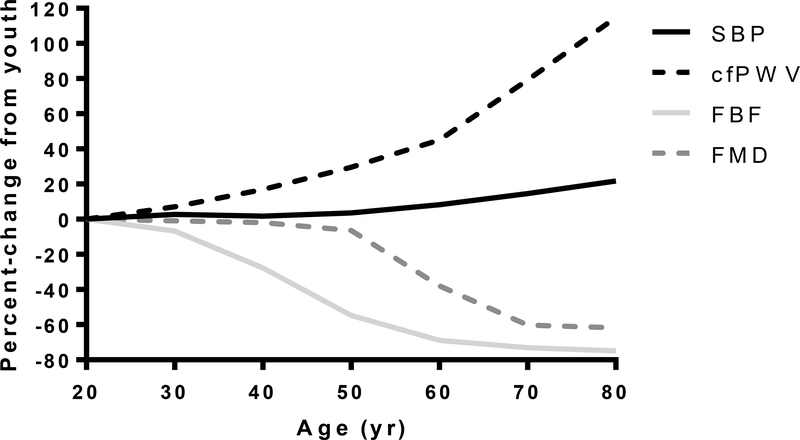
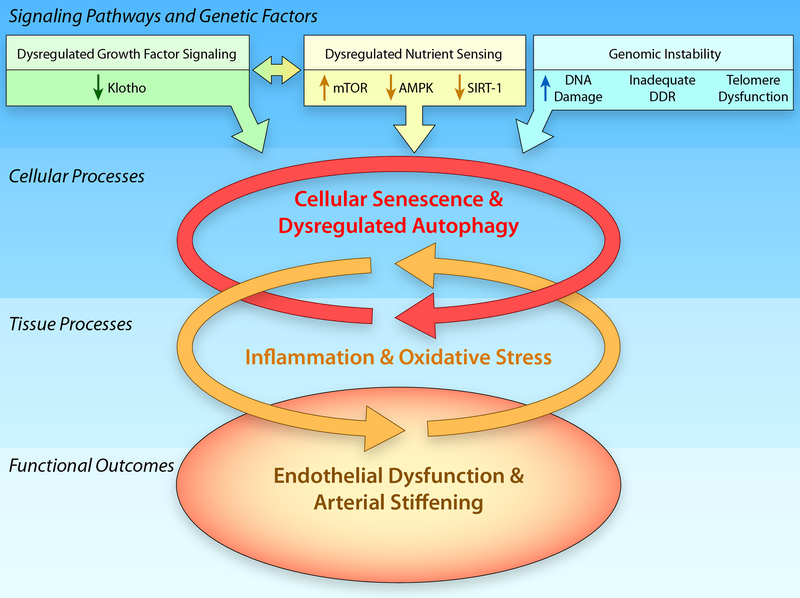
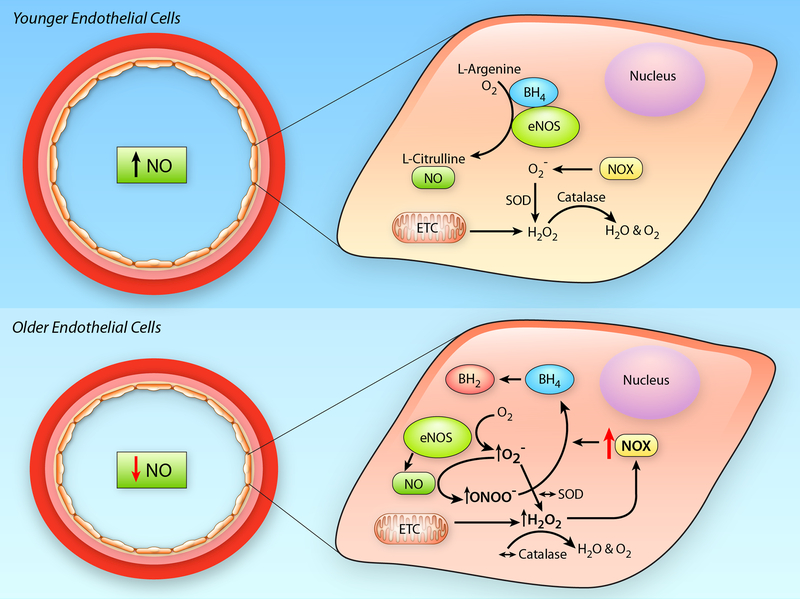
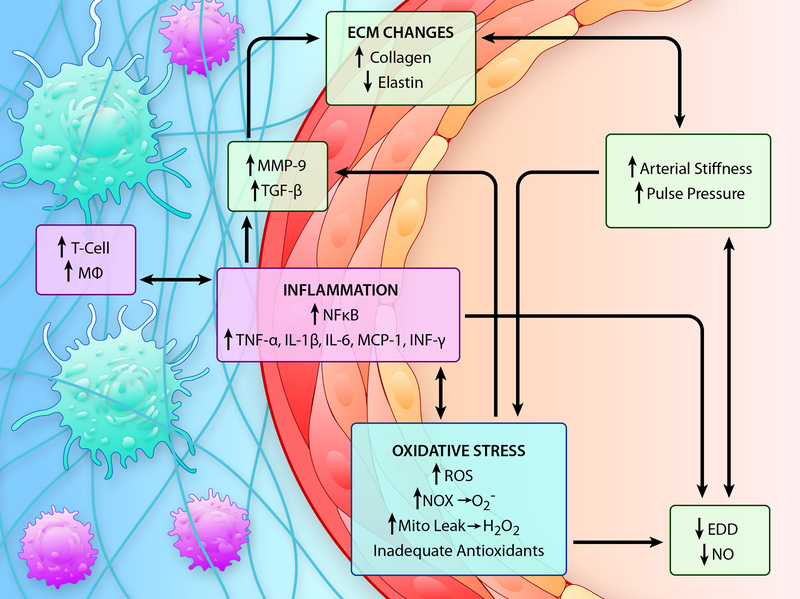
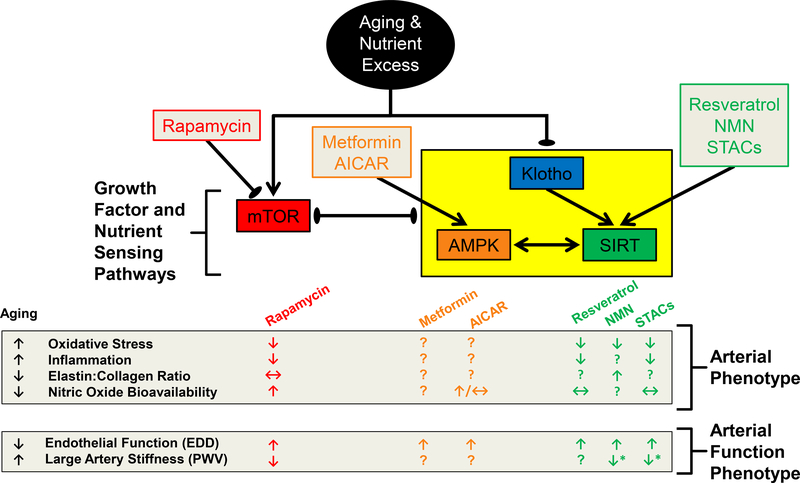
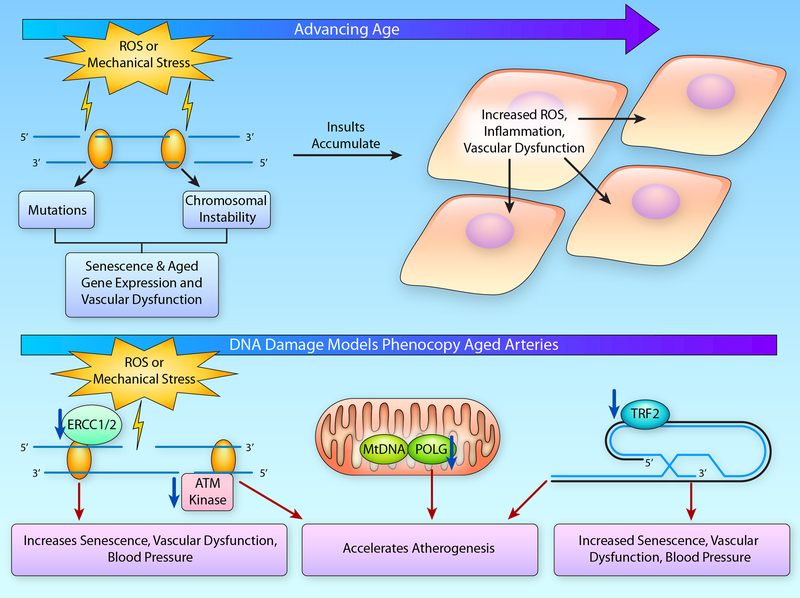
Advancing age promotes cardiovascular disease (CVD), the leading cause of death in the United States and many developed nations. Two major age-related arterial phenotypes, large elastic artery stiffening and endothelial dysfunction, are independent predictors of future CVD diagnosis and likely are responsible for the development of CVD in older adults. Not limited to traditional CVD, these age-related changes in the vasculature also contribute to other age-related diseases that influence mammalian health span and potential life span. This review explores mechanisms that influence age-related large elastic artery stiffening and endothelial dysfunction at the tissue level via inflammation and oxidative stress and at the cellular level via Klotho and energy-sensing pathways (AMPK [AMP-activated protein kinase], SIRT [sirtuins], and mTOR [mammalian target of rapamycin]). We also discuss how long-term calorie restriction-a health span- and life span-extending intervention-can prevent many of these age-related vascular phenotypes through the prevention of deleterious alterations in these mechanisms. Lastly, we discuss emerging novel mechanisms of vascular aging, including senescence and genomic instability within cells of the vasculature. As the population of older adults steadily expands, elucidating the cellular and molecular mechanisms of vascular dysfunction with age is critical to better direct appropriate and measured strategies that use pharmacological and lifestyle interventions to reduce risk of CVD within this population.
Keywords: aging; endothelium; inflammation; oxidative stress; telomere.
Figures
References
-
- Heron MP. Deaths: Leading causes for 2011. 2015
-
- Health, united states, 2010: With special feature on death and dying. Hyattsville (MD); 2011. - PubMed
-
- Lakatta EG, Levy D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part i: Aging arteries: A “set up” for vascular disease. Circulation. 2003;107:139–146 - PubMed
-
- Widlansky ME, Gokce N, Keaney JF, Jr., Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003;42:1149–1160 - PubMed
-
- Franklin SS, Khan SA, Wong ND, Larson MG, Levy D. Is pulse pressure useful in predicting risk for coronary heart disease? The framingham heart study. Circulation. 1999;100:354–360 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
