

Cellular and Molecular Mechanisms of Prion Disease
- PMID: 30355150
- PMCID: PMC9071098
- DOI: 10.1146/annurev-pathmechdis-012418-013109
Cellular and Molecular Mechanisms of Prion Disease
Abstract
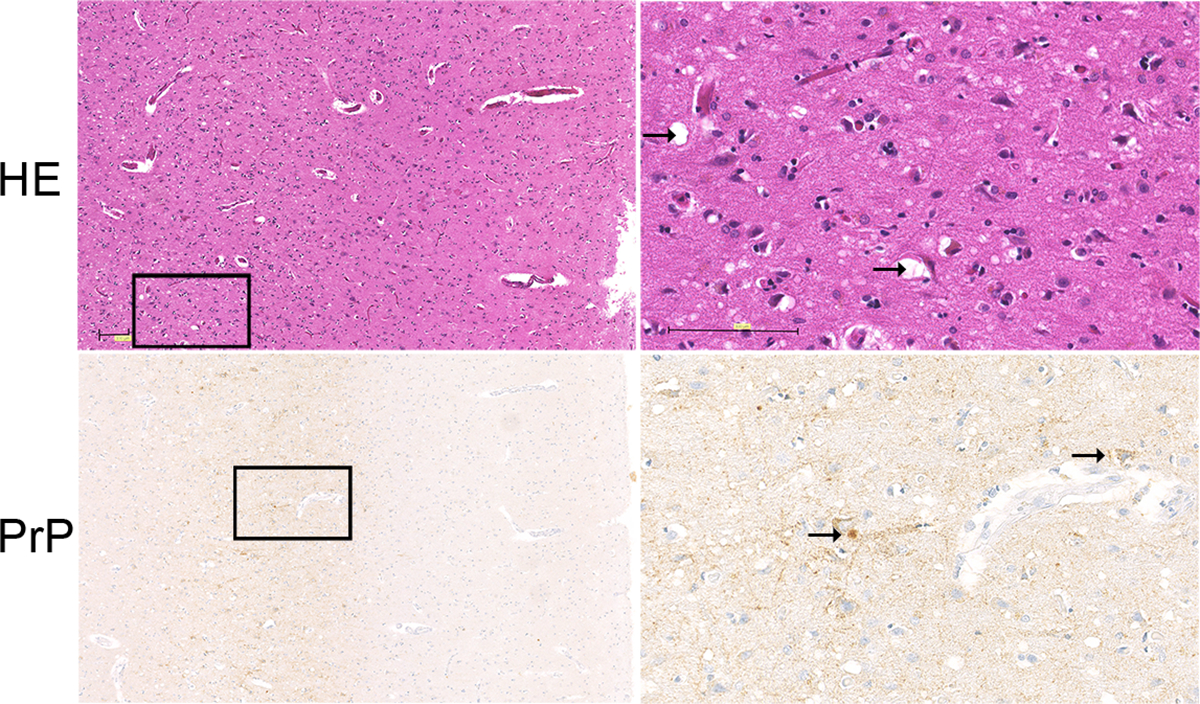
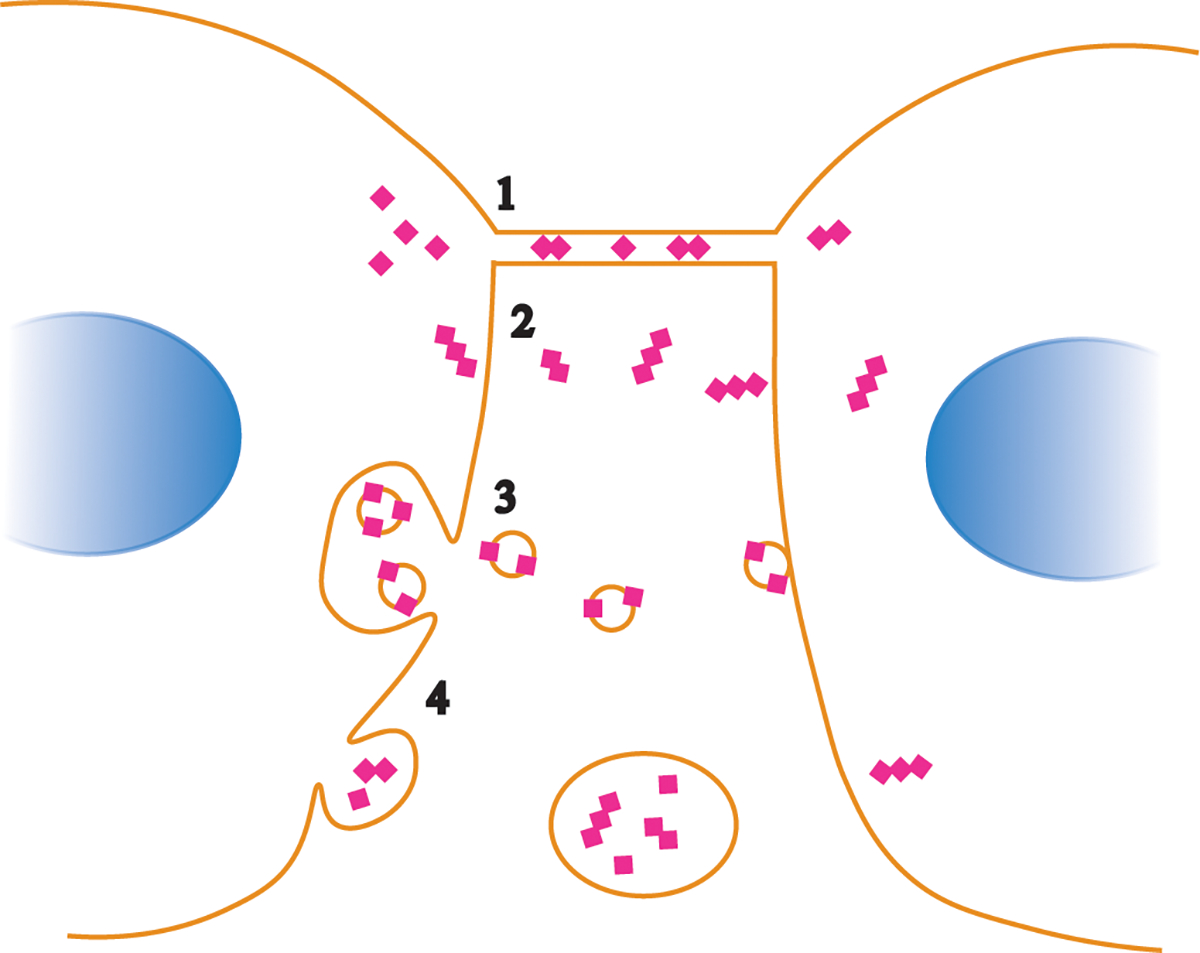
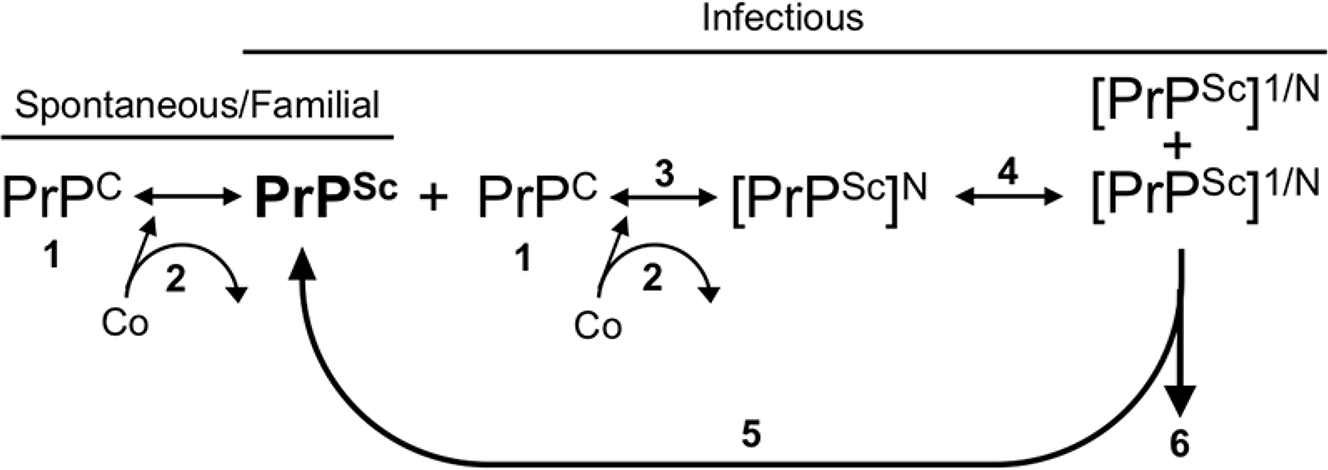
Prion diseases are rapidly progressive, incurable neurodegenerative disorders caused by misfolded, aggregated proteins known as prions, which are uniquely infectious. Remarkably, these infectious proteins have been responsible for widespread disease epidemics, including kuru in humans, bovine spongiform encephalopathy in cattle, and chronic wasting disease in cervids, the latter of which has spread across North America and recently appeared in Norway and Finland. The hallmark histopathological features include widespread spongiform encephalopathy, neuronal loss, gliosis, and deposits of variably sized aggregated prion protein, ranging from small, soluble oligomers to long, thin, unbranched fibrils, depending on the disease. Here, we explore recent advances in prion disease research, from the function of the cellular prion protein to the dysfunction triggering neurotoxicity, as well as mechanisms underlying prion spread between cells. We also highlight key findings that have revealed new therapeutic targets and consider unanswered questions for future research.
Keywords: amyloid; neurodegeneration; neurotoxicity; prion transmission; strains.
Figures
References
-
- Bolton DC, McKinley MP, Prusiner SB. 1982. Identification of a protein that purifies with the scrapie prion. Science 218: 1309–11 - PubMed
-
- Prusiner SB. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216: 136–44 - PubMed
-
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. 1998. Prion protein biology. Cell 93: 337–48 - PubMed
-
- Basler K, Oesch B, Scott M, Westaway D, Walchli M, Groth DF, McKinley MP, Prusiner SB, Weissmann C. 1986. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 46: 417–28 - PubMed
