

RNA Binding Proteins and the Pathogenesis of Frontotemporal Lobar Degeneration
- PMID: 30355151
- PMCID: PMC6731550
- DOI: 10.1146/annurev-pathmechdis-012418-012955
RNA Binding Proteins and the Pathogenesis of Frontotemporal Lobar Degeneration
Abstract
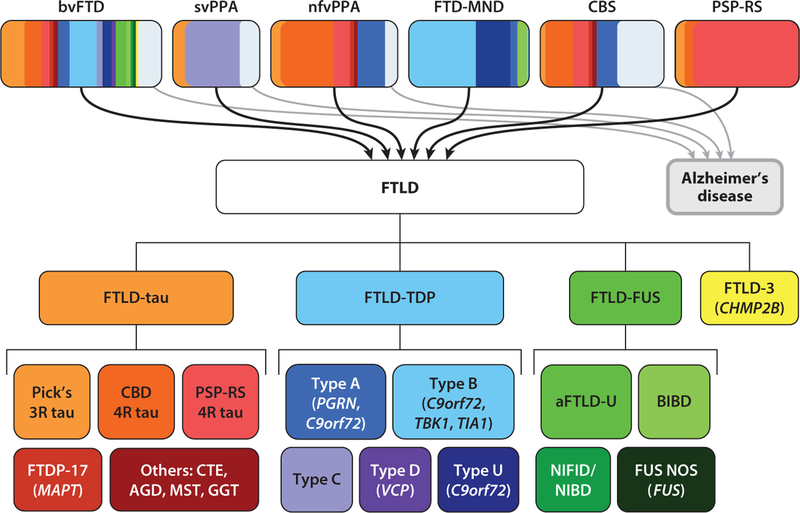
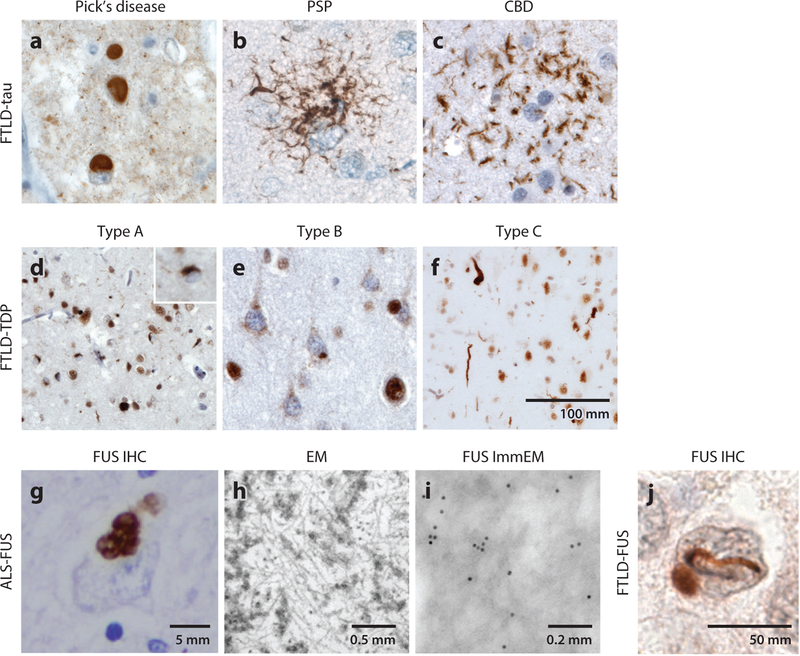
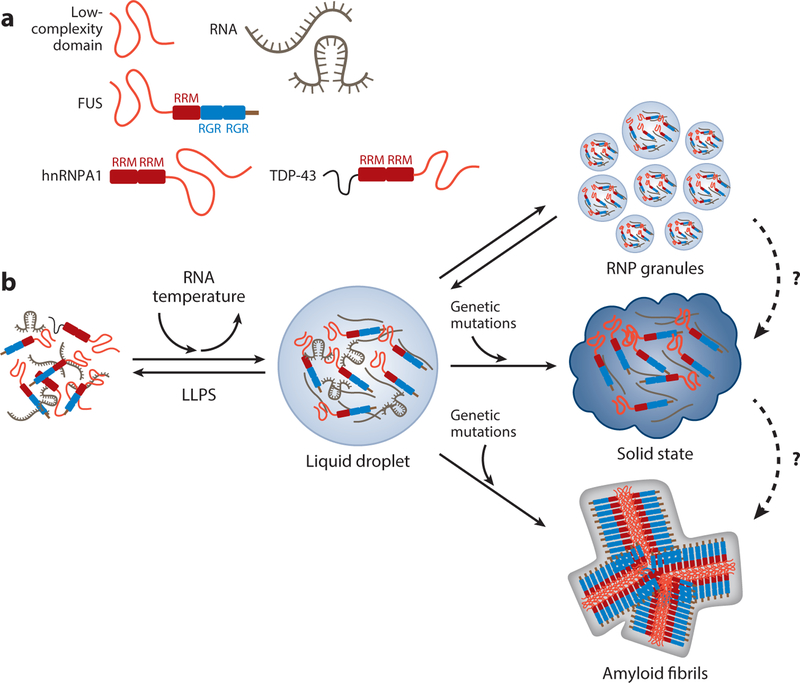
Frontotemporal dementia is a group of early onset dementia syndromes linked to underlying frontotemporal lobar degeneration (FTLD) pathology that can be classified based on the formation of abnormal protein aggregates involving tau and two RNA binding proteins, TDP-43 and FUS. Although elucidation of the mechanisms leading to FTLD pathology is in progress, recent advances in genetics and neuropathology indicate that a majority of FTLD cases with proteinopathy involving RNA binding proteins show highly congruent genotype-phenotype correlations. Specifically, recent studies have uncovered the unique properties of the low-complexity domains in RNA binding proteins that can facilitate liquid-liquid phase separation in the formation of membraneless organelles. Furthermore, there is compelling evidence that mutations in FTLD genes lead to dysfunction in diverse cellular pathways that converge on the endolysosomal pathway, autophagy, and neuroinflammation. Together, these results provide key mechanistic insights into the pathogenesis and potential therapeutic targets of FTLD.
Keywords: ALS; FTD; FTLD; FUS; RNA binding proteins; TDP-43; amyotrophic lateral sclerosis; frontotemporal dementia; frontotemporal lobar degeneration; hydrogels; liquid droplets; low-complexity domain.
Conflict of interest statement
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Figures

References
-
- Miller BL. 2013. Frontotemporal Dementia New York: Oxford Univ. Press
-
- Ratnavalli E, Brayne C, Dawson K, Hodges JR. 2002. The prevalence of frontotemporal dementia. Neurology 58:1615–21 - PubMed
-
- Pick A 1892. Uber die Beziehungen der senilen Hirnatrophie zur Aphasie. Prag. Med. Wochenschr 17:165–67
-
- Pick A 1904. Zur Symptomatologie der linksseitigen Schläfenlappenatrophie. Monatsschrift Psychiatr. Neurol 16:378–88
-
- Alzheimer A 1911. Uber eigenartige Krankheitsfälle des späteren Alters. Z. Gesamte Neurol. Psychiatr 4:356–85
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
