

Genomic comparison of Trypanosoma conorhini and Trypanosoma rangeli to Trypanosoma cruzi strains of high and low virulence
- PMID: 30355302
- PMCID: PMC6201504
- DOI: 10.1186/s12864-018-5112-0
Genomic comparison of Trypanosoma conorhini and Trypanosoma rangeli to Trypanosoma cruzi strains of high and low virulence
Abstract
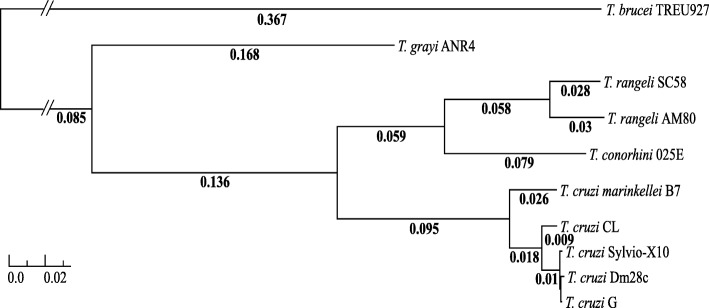
Background: Trypanosoma conorhini and Trypanosoma rangeli, like Trypanosoma cruzi, are kinetoplastid protist parasites of mammals displaying divergent hosts, geographic ranges and lifestyles. Largely nonpathogenic T. rangeli and T. conorhini represent clades that are phylogenetically closely related to the T. cruzi and T. cruzi-like taxa and provide insights into the evolution of pathogenicity in those parasites. T. rangeli, like T. cruzi is endemic in many Latin American countries, whereas T. conorhini is tropicopolitan. T. rangeli and T. conorhini are exclusively extracellular, while T. cruzi has an intracellular stage in the mammalian host.
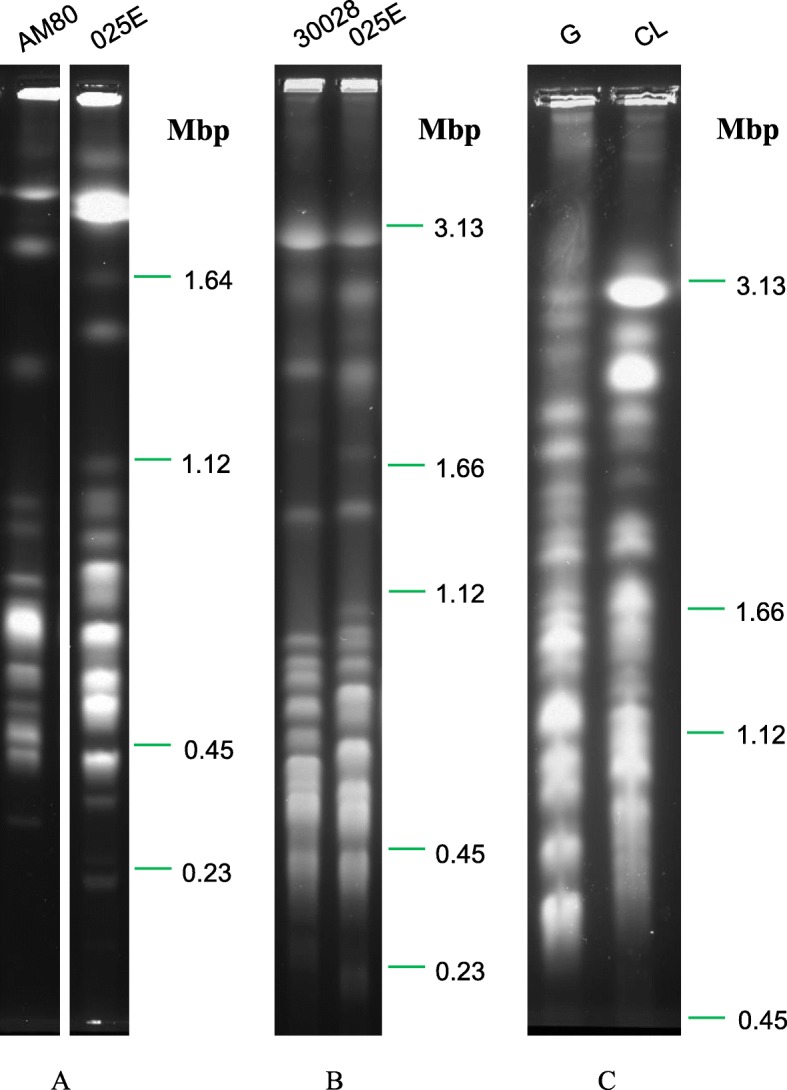
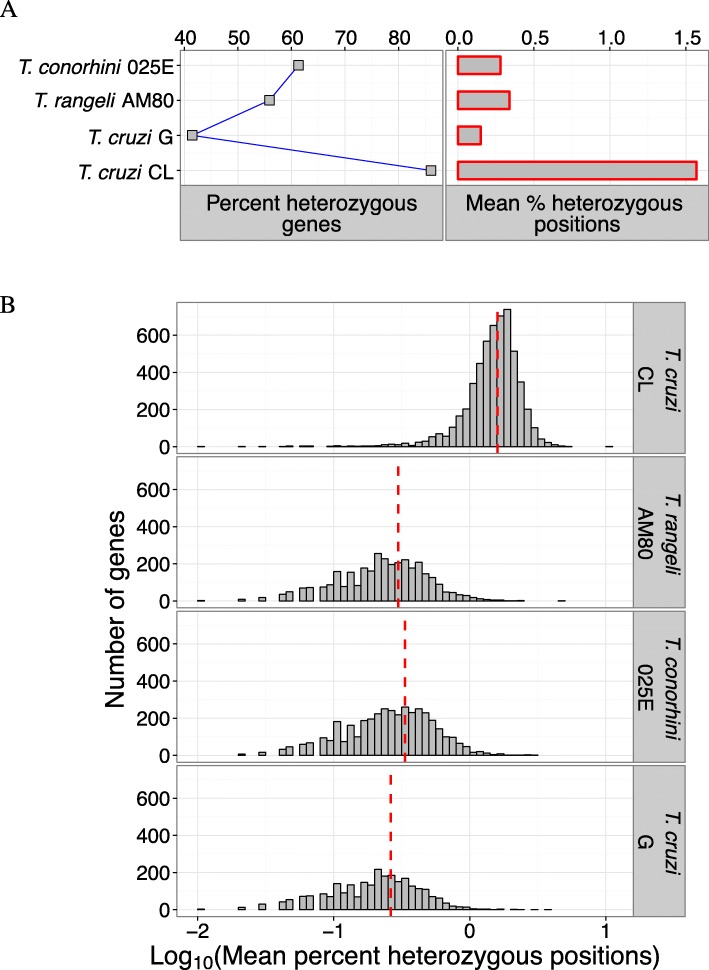
Results: Here we provide the first comprehensive sequence analysis of T. rangeli AM80 and T. conorhini 025E, and provide a comparison of their genomes to those of T. cruzi G and T. cruzi CL, respectively members of T. cruzi lineages TcI and TcVI. We report de novo assembled genome sequences of the low-virulent T. cruzi G, T. rangeli AM80, and T. conorhini 025E ranging from ~ 21-25 Mbp, with ~ 10,000 to 13,000 genes, and for the highly virulent and hybrid T. cruzi CL we present a ~ 65 Mbp in-house assembled haplotyped genome with ~ 12,500 genes per haplotype. Single copy orthologs of the two T. cruzi strains exhibited ~ 97% amino acid identity, and ~ 78% identity to proteins of T. rangeli or T. conorhini. Proteins of the latter two organisms exhibited ~ 84% identity. T. cruzi CL exhibited the highest heterozygosity. T. rangeli and T. conorhini displayed greater metabolic capabilities for utilization of complex carbohydrates, and contained fewer retrotransposons and multigene family copies, i.e. trans-sialidases, mucins, DGF-1, and MASP, compared to T. cruzi.
Conclusions: Our analyses of the T. rangeli and T. conorhini genomes closely reflected their phylogenetic proximity to the T. cruzi clade, and were largely consistent with their divergent life cycles. Our results provide a greater context for understanding the life cycles, host range expansion, immunity evasion, and pathogenesis of these trypanosomatids.
Keywords: Comparative genomics; Genome sequencing; Trypanosomatids.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no completing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures


References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
