

NanoPARE: parallel analysis of RNA 5' ends from low-input RNA
- PMID: 30355603
- PMCID: PMC6280765
- DOI: 10.1101/gr.239202.118
NanoPARE: parallel analysis of RNA 5' ends from low-input RNA
Abstract
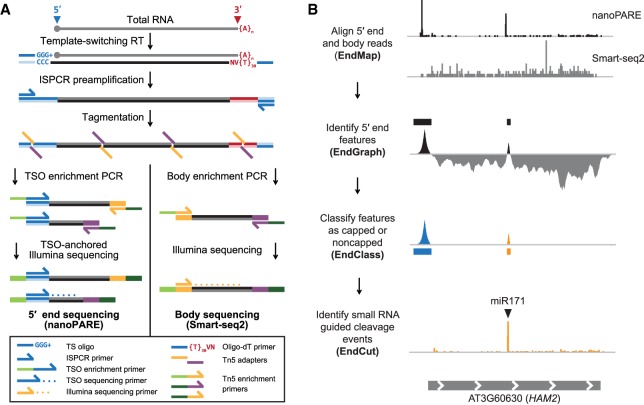
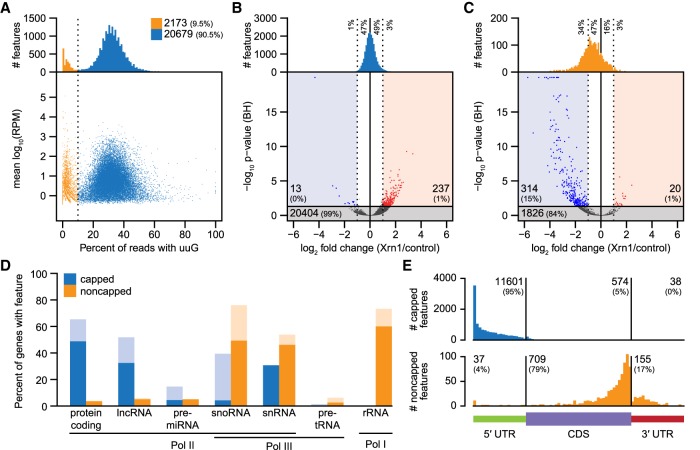
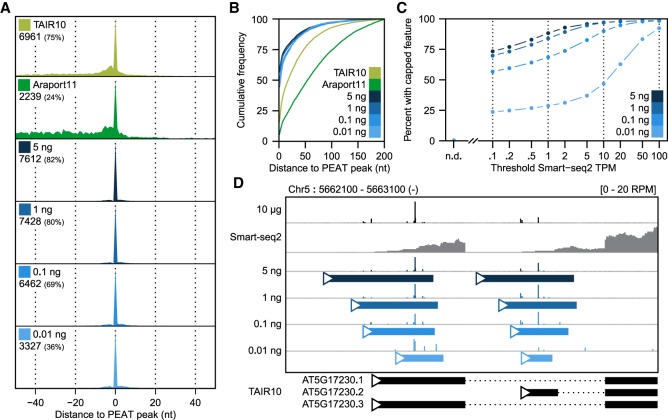
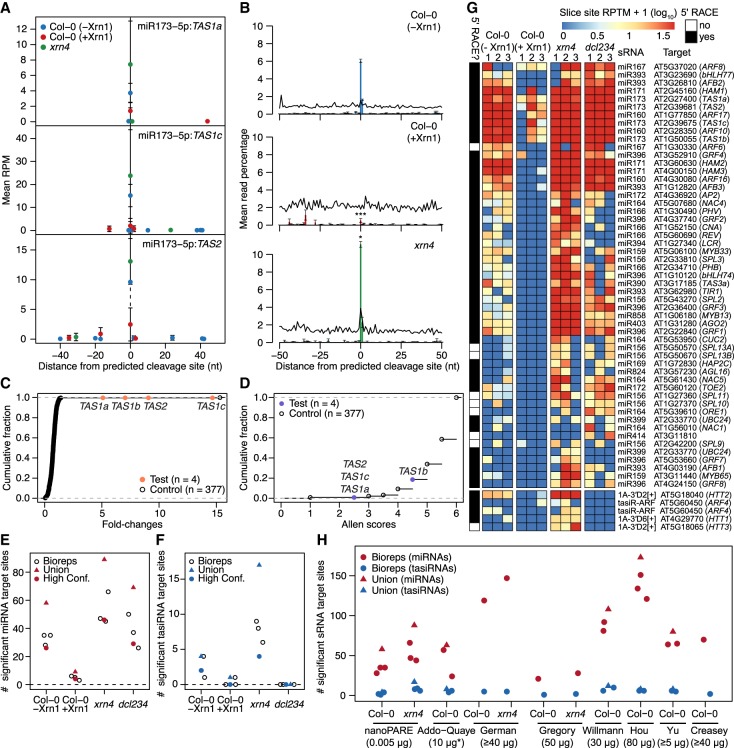
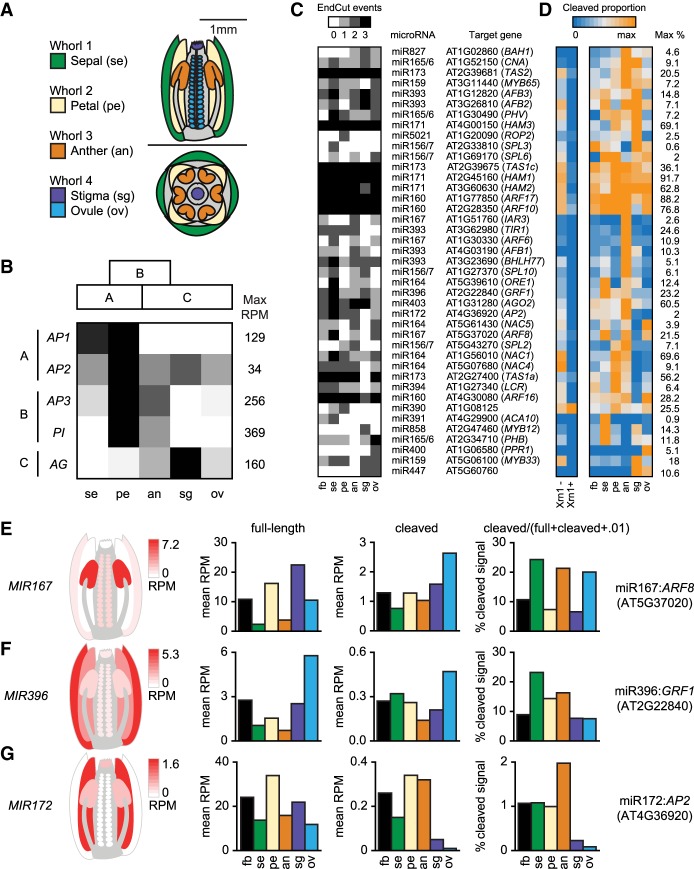
Diverse RNA 5' ends are generated through both transcriptional and post-transcriptional processes. These important modes of gene regulation often vary across cell types and can contribute to the diversification of transcriptomes and thus cellular differentiation. Therefore, the identification of primary and processed 5' ends of RNAs is important for their functional characterization. Methods have been developed to profile either RNA 5' ends from primary transcripts or the products of RNA degradation genome-wide. However, these approaches either require high amounts of starting RNA or are performed in the absence of paired gene-body mRNA-seq data. This limits current efforts in RNA 5' end annotation to whole tissues and can prevent accurate RNA 5' end classification due to biases in the data sets. To enable the accurate identification and precise classification of RNA 5' ends from standard and low-input RNA, we developed a next-generation sequencing-based method called nanoPARE and associated software. By integrating RNA 5' end information from nanoPARE with gene-body mRNA-seq data from the same RNA sample, our method enables the identification of transcription start sites at single-nucleotide resolution from single-cell levels of total RNA, as well as small RNA-mediated cleavage events from at least 10,000-fold less total RNA compared to conventional approaches. NanoPARE can therefore be used to accurately profile transcription start sites, noncapped RNA 5' ends, and small RNA targeting events from individual tissue types. As a proof-of-principle, we utilized nanoPARE to improve Arabidopsis thaliana RNA 5' end annotations and quantify microRNA-mediated cleavage events across five different flower tissues.
© 2018 Schon et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
Riboswitch discovery by combining RNA-seq and genome-wide identification of transcriptional start sites.Methods Enzymol. 2014;549:3-27. doi: 10.1016/B978-0-12-801122-5.00001-5. Methods Enzymol. 2014. PMID: 25432742
-
RNA Sequencing for Transcript 5'-End Mapping in Mycobacteria.Methods Mol Biol. 2021;2314:513-531. doi: 10.1007/978-1-0716-1460-0_22. Methods Mol Biol. 2021. PMID: 34235668
-
Capturing 5' and 3' native ends of mRNAs concurrently with Akron sequencing.Nat Protoc. 2019 May;14(5):1578-1602. doi: 10.1038/s41596-019-0151-9. Epub 2019 Apr 10. Nat Protoc. 2019. PMID: 30971782
-
The use of high-throughput sequencing methods for plant microRNA research.RNA Biol. 2015;12(7):709-19. doi: 10.1080/15476286.2015.1053686. RNA Biol. 2015. PMID: 26016494 Free PMC article. Review.
-
Challenges in identifying mRNA transcript starts and ends from long-read sequencing data.Genome Res. 2024 Nov 20;34(11):1719-1734. doi: 10.1101/gr.279559.124. Genome Res. 2024. PMID: 39567236 Free PMC article. Review.
Cited by
-
Global approaches for profiling transcription initiation.Cell Rep Methods. 2021 Sep 27;1(5):100081. doi: 10.1016/j.crmeth.2021.100081. Epub 2021 Sep 16. Cell Rep Methods. 2021. PMID: 34632443 Free PMC article. Review.
-
Bookend: precise transcript reconstruction with end-guided assembly.Genome Biol. 2022 Jun 29;23(1):143. doi: 10.1186/s13059-022-02700-3. Genome Biol. 2022. PMID: 35768836 Free PMC article.
-
NATpare: a pipeline for high-throughput prediction and functional analysis of nat-siRNAs.Nucleic Acids Res. 2020 Jul 9;48(12):6481-6490. doi: 10.1093/nar/gkaa448. Nucleic Acids Res. 2020. PMID: 32463462 Free PMC article.
-
Widespread premature transcription termination of Arabidopsis thaliana NLR genes by the spen protein FPA.Elife. 2021 Apr 27;10:e65537. doi: 10.7554/eLife.65537. Elife. 2021. PMID: 33904405 Free PMC article.
-
PAMP-triggered genetic reprogramming involves widespread alternative transcription initiation and an immediate transcription factor wave.Plant Cell. 2022 Jul 4;34(7):2615-2637. doi: 10.1093/plcell/koac108. Plant Cell. 2022. PMID: 35404429 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials