

What is an archaeon and are the Archaea really unique?
- PMID: 30357005
- PMCID: PMC6196074
- DOI: 10.7717/peerj.5770
What is an archaeon and are the Archaea really unique?
Abstract
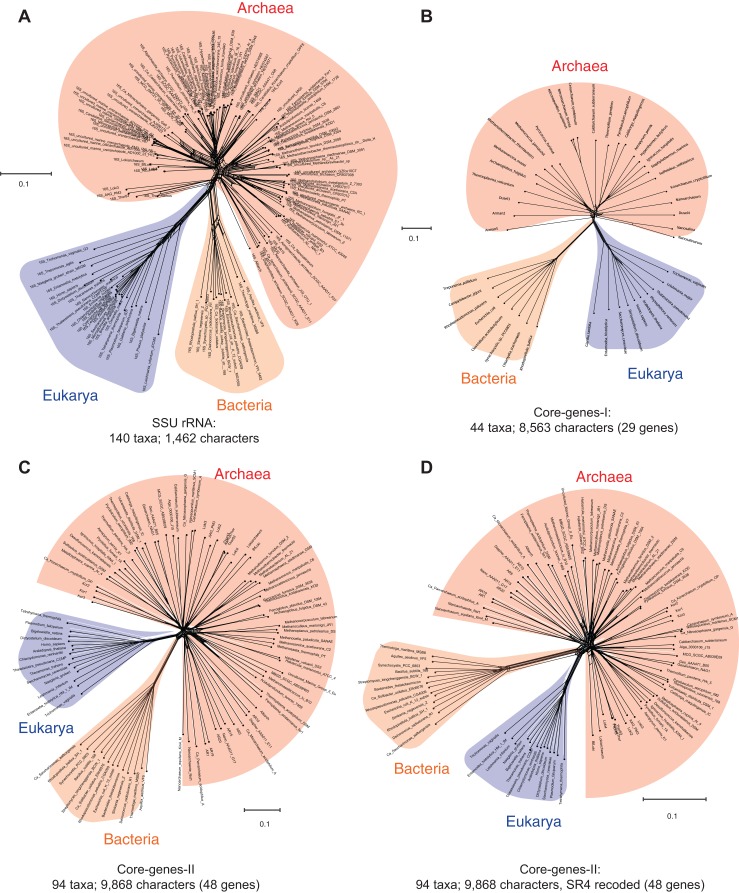
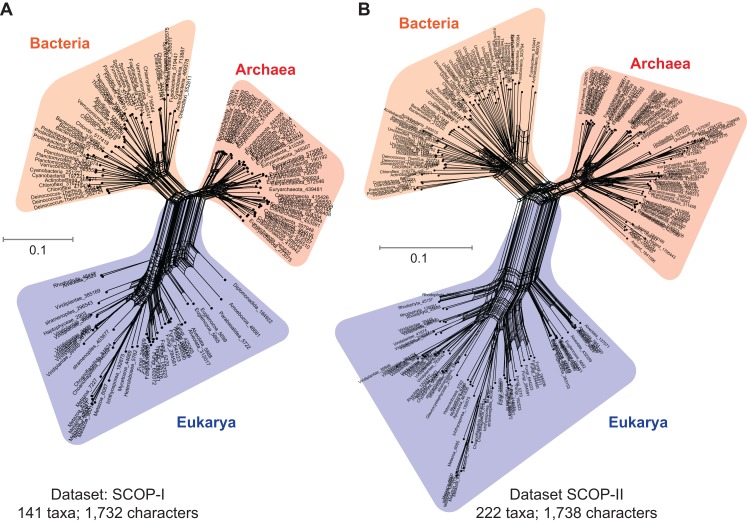
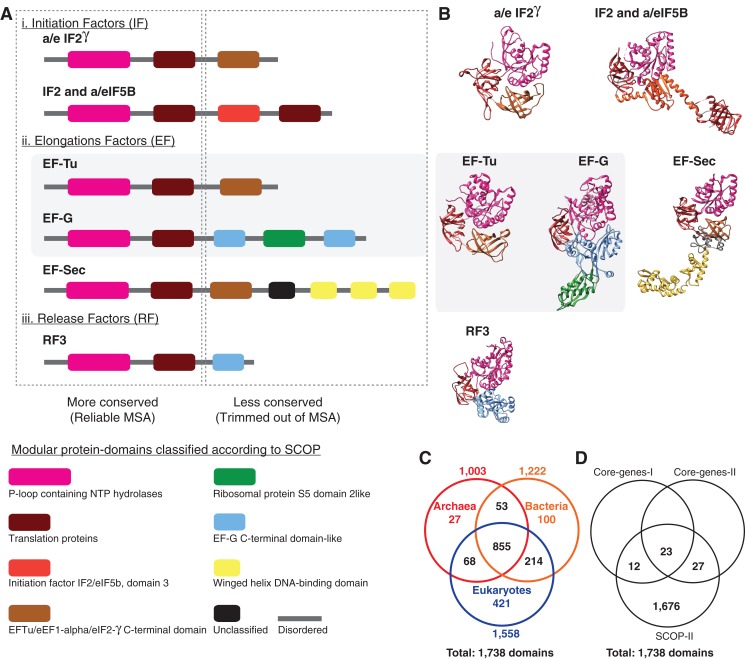
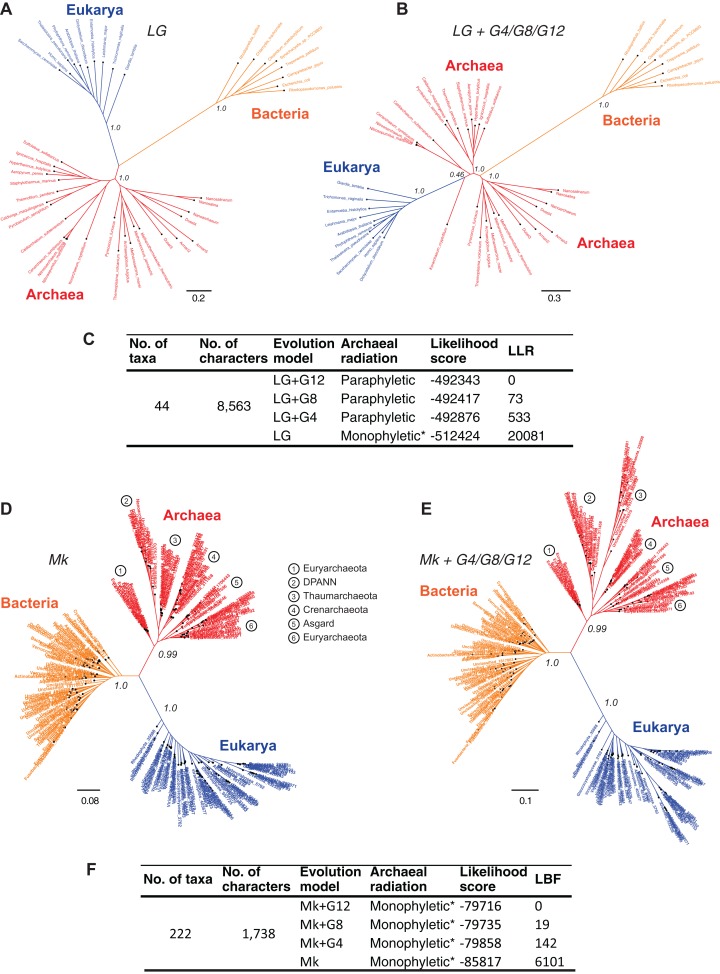
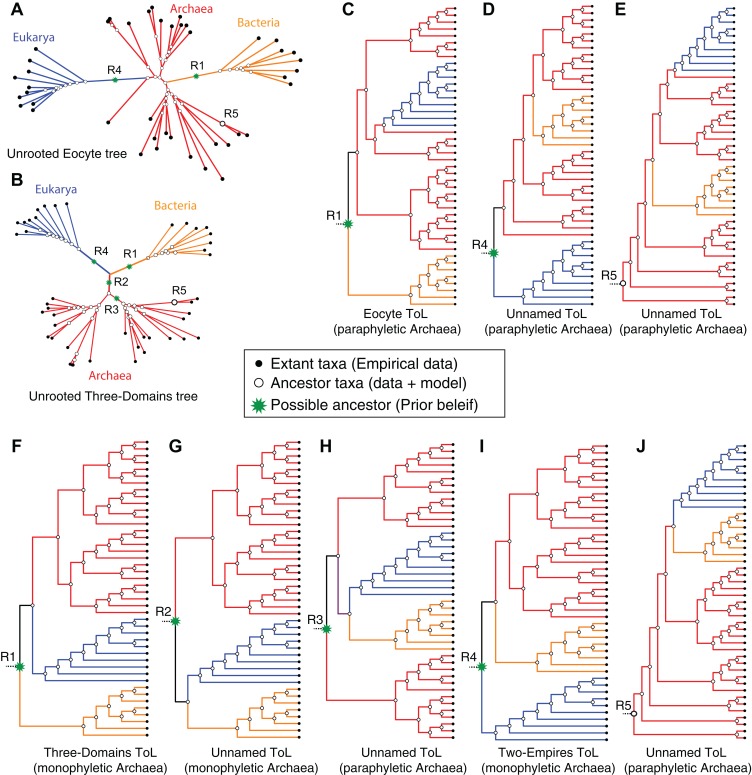
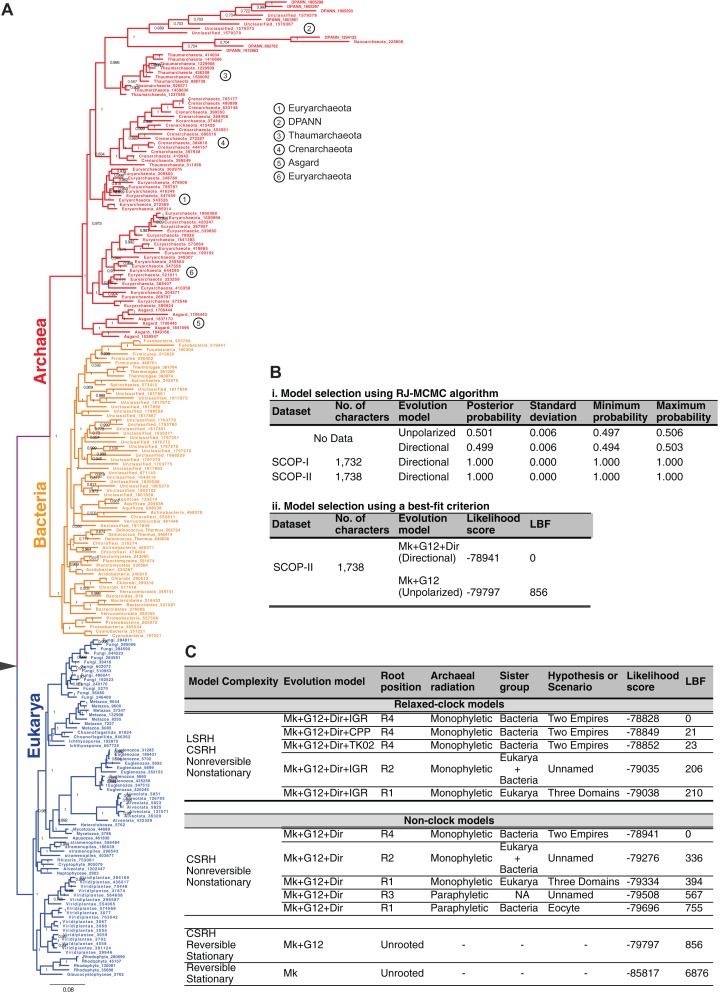
The recognition of the group Archaea as a major branch of the tree of life (ToL) prompted a new view of the evolution of biodiversity. The genomic representation of archaeal biodiversity has since significantly increased. In addition, advances in phylogenetic modeling of multi-locus datasets have resolved many recalcitrant branches of the ToL. Despite the technical advances and an expanded taxonomic representation, two important aspects of the origins and evolution of the Archaea remain controversial, even as we celebrate the 40th anniversary of the monumental discovery. These issues concern (i) the uniqueness (monophyly) of the Archaea, and (ii) the evolutionary relationships of the Archaea to the Bacteria and the Eukarya; both of these are relevant to the deep structure of the ToL. To explore the causes for this persistent ambiguity, I examine multiple datasets and different phylogenetic approaches that support contradicting conclusions. I find that the uncertainty is primarily due to a scarcity of information in standard datasets-universal core-genes datasets-to reliably resolve the conflicts. These conflicts can be resolved efficiently by comparing patterns of variation in the distribution of functional genomic signatures, which are less diffused unlike patterns of primary sequence variation. Relatively lower heterogeneity in distribution patterns minimizes uncertainties and supports statistically robust phylogenetic inferences, especially of the earliest divergences of life. This case study further highlights the limitations of primary sequence data in resolving difficult phylogenetic problems, and raises questions about evolutionary inferences drawn from the analyses of sequence alignments of a small set of core genes. In particular, the findings of this study corroborate the growing consensus that reversible substitution mutations may not be optimal phylogenetic markers for resolving early divergences in the ToL, nor for determining the polarity of evolutionary transitions across the ToL.
Keywords: Archaea; Asgard; Chimeric genome; Clade; Directional evolution; Genome fusion; Non-stationary; Phylogenomics; Rooting; Tree of life.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, Thomas BC, Singh A, Wilkins MJ, Karaoz U, Brodie EL, Williams KH, Hubbard SS, Banfield JF. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nature Communications. 2016;7:13219. doi: 10.1038/ncomms13219. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
