

N-terminal extension in cardiac myosin-binding protein C regulates myofilament binding
- PMID: 30359561
- PMCID: PMC6366860
- DOI: 10.1016/j.yjmcc.2018.10.009
N-terminal extension in cardiac myosin-binding protein C regulates myofilament binding
Abstract
Rationale: Mutations in the gene encoding the sarcomeric protein cardiac myosin-binding protein C (cMyBP-C) are a leading cause of hypertrophic cardiomyopathy (HCM). Mouse models targeting cMyBP-C and use of recombinant proteins have been effective in studying its roles in contractile function and disease. Surprisingly, while the N-terminus of cMyBP-C is important to regulate myofilament binding and contains many HCM mutations, an incorrect sequence, lacking the N-terminal 8 amino acids has been used in many studies.
Objectives: To determine the N-terminal cMyBP-C sequences in ventricles and investigate the roles of species-specific differences in cMyBP-C on myofilament binding.
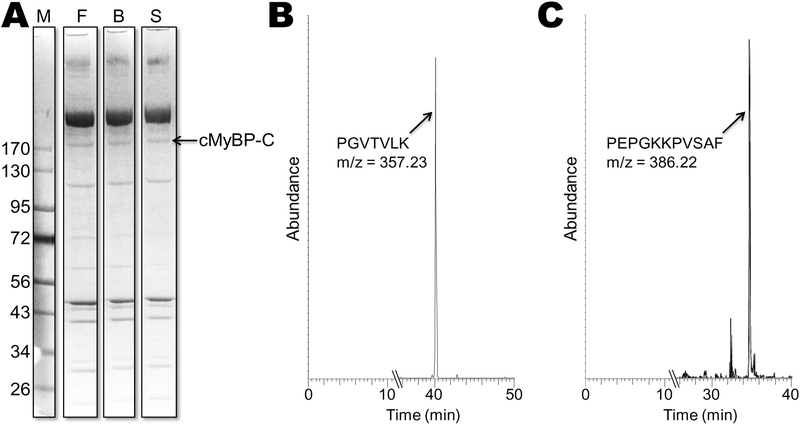
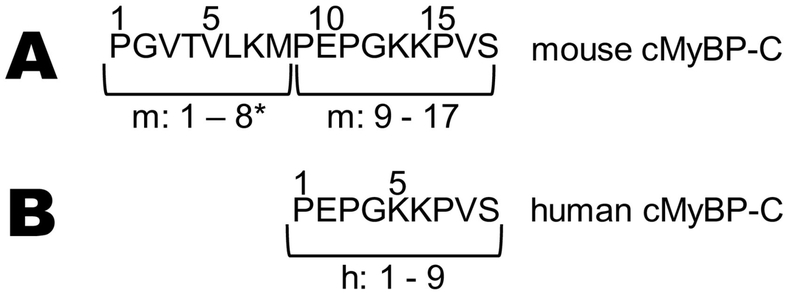
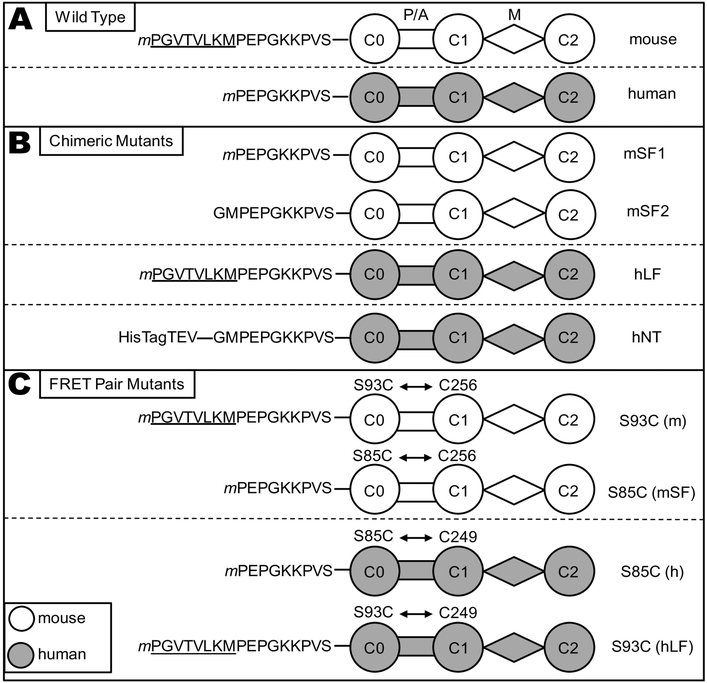
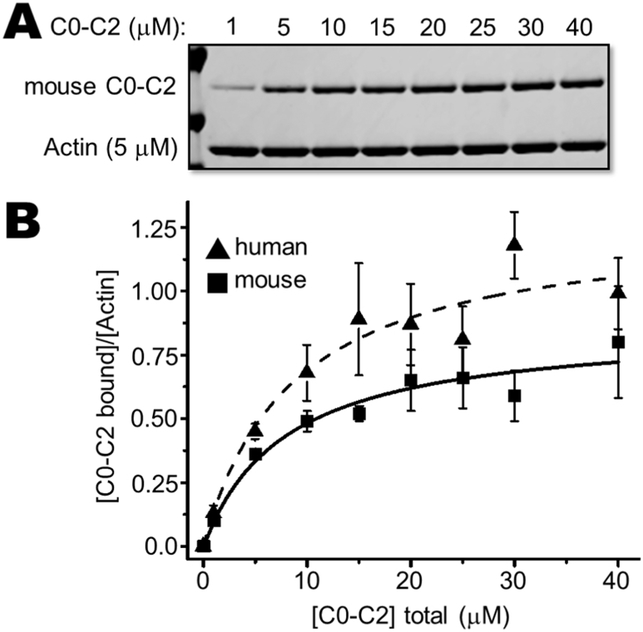
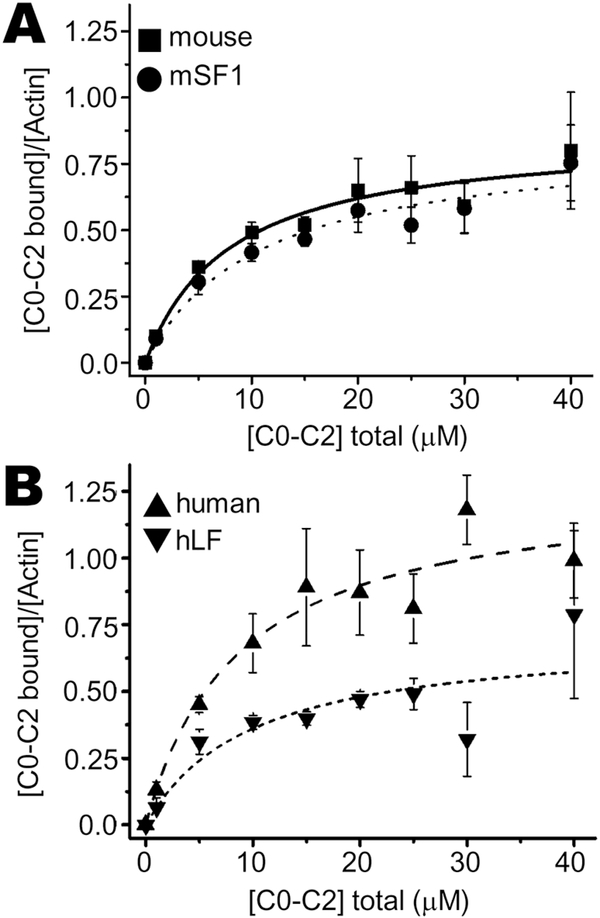
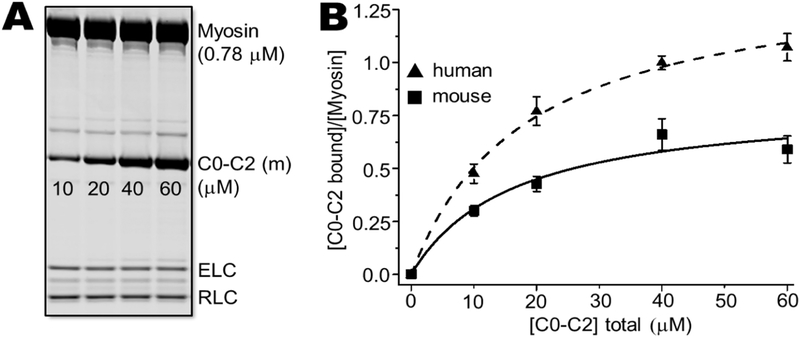
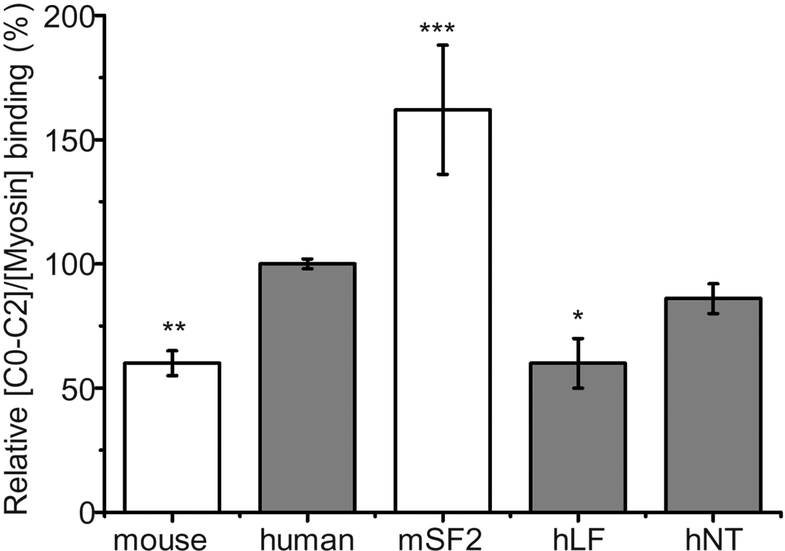
Methods and results: We determined cMyBP-C sequences in mouse and human by inspecting available sequence databases. N-terminal differences were confirmed using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Cosedimentation assays with actin or myosin were used to examine binding in mouse, human and chimeric fusion proteins of cMyBP-C. Time-resolved FRET (TR-FRET) with site-directed probes on cMyBP-C was employed to measure structural dynamics. LC-MS/MS supported the sequencing data that mouse cMyBP-C contains an eight-residue N-terminal extension (NTE) not found in human. Cosedimentation assays revealed that cardiac myosin binding was strongly influenced by the presence of the NTE, which reduced binding by 60%. 75% more human C0-C2 than mouse bound to myosin. Actin binding of mouse C0-C2 was not affected by the NTE. 50% more human C0-C2 than mouse bound to actin. TR-FRET indicates that the NTE did not significantly affect structural dynamics across domains C0 and C1.
Conclusions: Our functional results are consistent with the idea that cardiac myosin binding of N-terminal cMyBP-C is reduced in the mouse protein due to the presence of the NTE, which is proposed to interfere with myosin regulatory light chain (RLC) binding. The NTE is a critical component of mouse cMyBP-C, and should be considered in extrapolation of studies to cMyBP-C and HCM mechanisms in human.
Keywords: Cardiac myosin-binding protein C; Contractile proteins; Myofilament; Myosin; N-terminal extension; Regulation.
Copyright © 2018 The Authors. Published by Elsevier Ltd.. All rights reserved.
Conflict of interest statement
Figures
References
-
- Semsarian C, Ingles J, Maron MS, Maron BJ, New perspectives on the prevalence of hypertrophic cardiomyopathy, J Am Coll Cardiol 65(12) (2015) 1249–54. - PubMed
-
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW, G. American College of Cardiology Foundation/American Heart Association Task Force on Practice, 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons, J Am Coll Cardiol 58(25) (2011) e212–60. - PubMed
-
- Theis JL, Bos JM, Theis JD, Miller DV, Dearani JA, Schaff HV, Gersh BJ, Ommen SR, Moss RL, Ackerman MJ, Expression patterns of cardiac myofilament proteins: genomic and protein analysis of surgical myectomy tissue from patients with obstructive hypertrophic cardiomyopathy, Circ Heart Fail 2(4) (2009) 325–33. - PMC - PubMed
-
- Xu Q, Dewey S, Nguyen S, Gomes AV, Malignant and benign mutations in familial cardiomyopathies: insights into mutations linked to complex cardiovascular phenotypes, J Mol Cell Cardiol 48(5) (2010) 899–909. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
