

The Clinical Biology of Cystic Fibrosis Transmembrane Regulator Protein: Its Role and Function in Extrapulmonary Disease
- PMID: 30359614
- PMCID: PMC6414788
- DOI: 10.1016/j.chest.2018.10.006
The Clinical Biology of Cystic Fibrosis Transmembrane Regulator Protein: Its Role and Function in Extrapulmonary Disease
Abstract
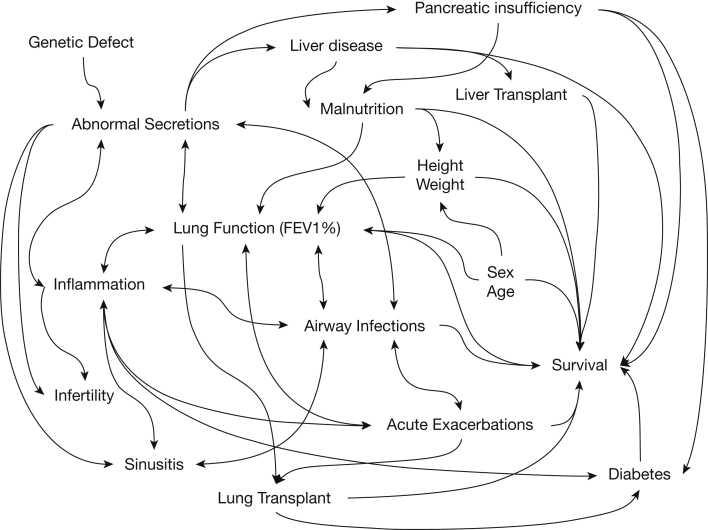
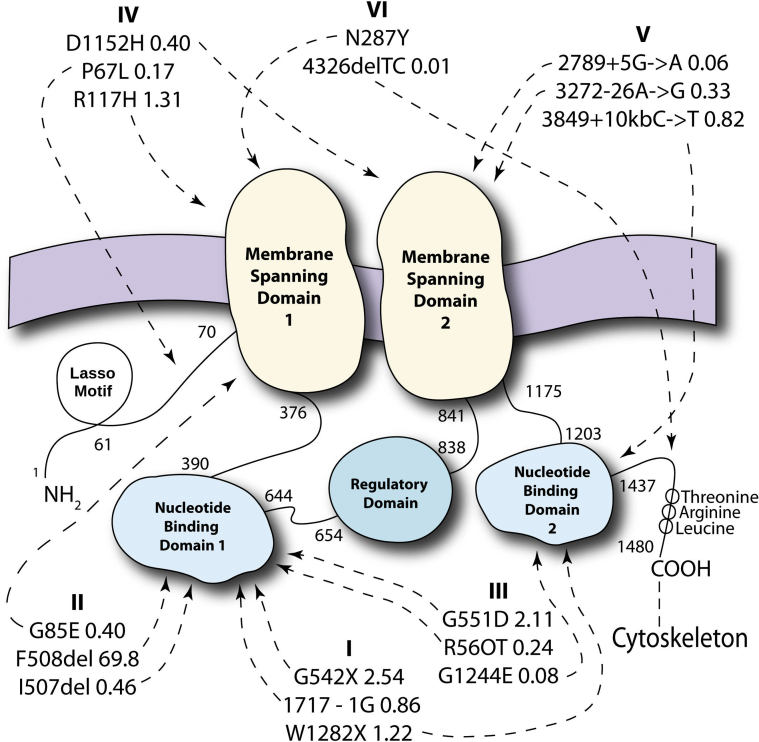
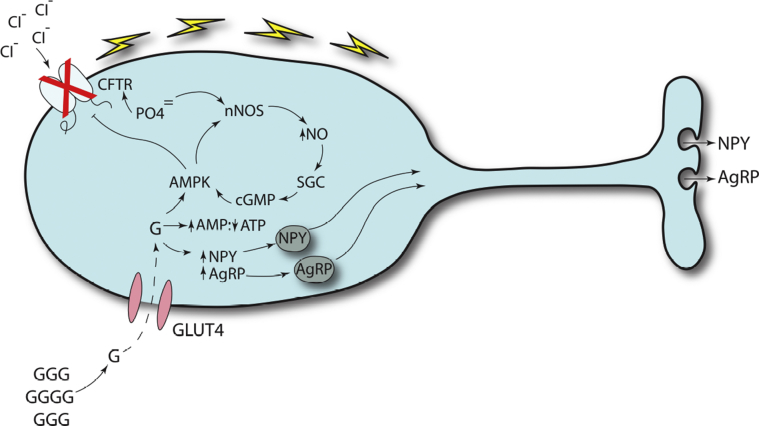
Normal cystic fibrosis (CF) transmembrane regulator (CFTR) protein has multiple functions in health and disease. Many mutations in the CFTR gene produce abnormal or absent protein. CFTR protein dysfunction underlies the classic CF phenotype of progressive pulmonary and GI pathology but may underlie diseases not usually associated with CF. This review highlights selected extrapulmonary disease that may be associated with abnormal CFTR. Increasing survival in CF is associated with increasing incidence of diseases associated with aging. CFTR dysfunction in older individuals may have novel effects on glucose metabolism, control of insulin release, regulation of circadian rhythm, and cancer cell pathophysiology. In individuals who have cancers with acquired CFTR suppression, their tumors may more likely exhibit rapid expansion, epithelial-to-mesenchymal transformation, abnormally reduced apoptosis, and increased metastatic potential. The new modulators of CFTR protein synthesis could facilitate the additional exploration needed to better understand the unfolding clinical biology of CFTR in human disease, even as they revolutionize treatment of patients with CF.
Keywords: cancer; circadian rhythm; cystic fibrosis transmembrane regulator; hypoglycemia; sleep.
Copyright © 2018 American College of Chest Physicians. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Elborn J.S. Cystic fibrosis. Lancet. 2016;388(10059):2519–2531. - PubMed
-
- Habib A.R., Buxton J.A., Singer J., Wilcox P.G., Javer A.R., Quon B.S. Association between chronic rhinosinusitis and health-related quality of life in adults with cystic fibrosis. Ann Am Thorac Soc. 2015;12(8):1163–1169. - PubMed
