

Whole genome sequencing for investigations of meningococcal outbreaks in the United States: a retrospective analysis
- PMID: 30361650
- PMCID: PMC6202316
- DOI: 10.1038/s41598-018-33622-5
Whole genome sequencing for investigations of meningococcal outbreaks in the United States: a retrospective analysis
Abstract
Although rare in the U.S., outbreaks due to Neisseria meningitidis do occur. Rapid, early outbreak detection is important for timely public health response. In this study, we characterized U.S. meningococcal isolates (N = 201) from 15 epidemiologically defined outbreaks (2009-2015) along with temporally and geographically matched sporadic isolates using multilocus sequence typing, pulsed-field gel electrophoresis (PFGE), and six whole genome sequencing (WGS) based methods. Recombination-corrected maximum likelihood (ML) and Bayesian phylogenies were reconstructed to identify genetically related outbreak isolates. All WGS analysis methods showed high degree of agreement and distinguished isolates with similar or indistinguishable PFGE patterns, or the same strain genotype. Ten outbreaks were caused by a single strain; 5 were due to multiple strains. Five sporadic isolates were phylogenetically related to 2 outbreaks. Analysis of 9 outbreaks using timed phylogenies identified the possible origin and estimated the approximate time that the most recent common ancestor emerged for outbreaks analyzed. U.S. meningococcal outbreaks were caused by single- or multiple-strain introduction, with organizational outbreaks mainly caused by a clonal strain and community outbreaks by divergent strains. WGS can infer linkage of meningococcal cases when epidemiological links are uncertain. Accurate identification of outbreak-associated cases requires both WGS typing and epidemiological data.
Conflict of interest statement
The authors declare no competing interests.
Figures
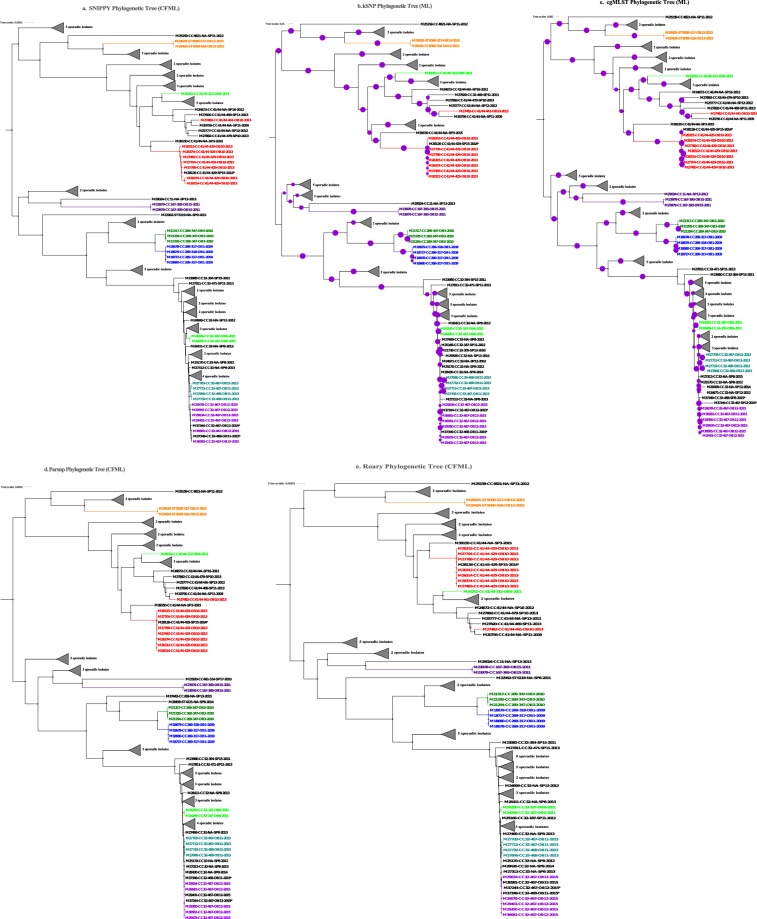
 represented 100% bootstrap support. Bootstrap support was estimated only for kSNP and cgMLST ML phylogenetic trees; the Bayesian recombination-adjusted phylogenies from ClonalFrameML do not estimate bootstrap values. Isolate label contains isolate ID, CC, PFGE pattern, outbreak or sporadic isolates, and year. Geographically matched outbreak and sporadic isolates are indicated by same number following “SP” or “OB” in label. Each colored branch and isolate label represents a different outbreak.
represented 100% bootstrap support. Bootstrap support was estimated only for kSNP and cgMLST ML phylogenetic trees; the Bayesian recombination-adjusted phylogenies from ClonalFrameML do not estimate bootstrap values. Isolate label contains isolate ID, CC, PFGE pattern, outbreak or sporadic isolates, and year. Geographically matched outbreak and sporadic isolates are indicated by same number following “SP” or “OB” in label. Each colored branch and isolate label represents a different outbreak.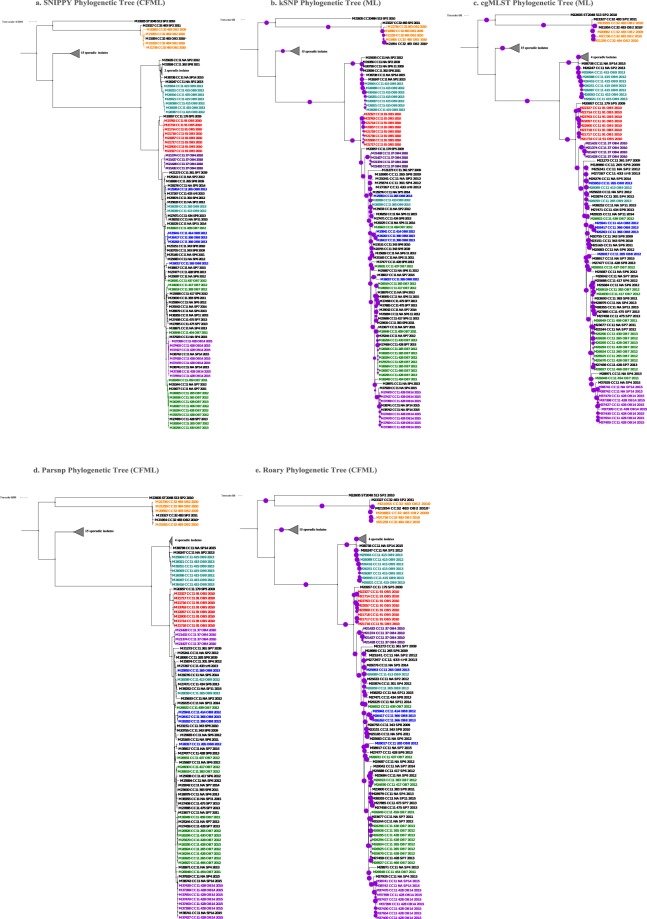
 represented 100% bootstrap support. Bootstrap support was estimated only for kSNP and cgMLST ML phylogenetic trees; the Bayesian recombination-adjusted phylogenies from ClonalFrameML do not estimate bootstrap values. Isolate label contains isolate ID, CC, PFGE pattern, outbreak or sporadic isolates, and year. Geographically matched sporadic and outbreak isolates are indicated by the same number following “SP” or “OB” in label. M26263 and M26417 were isolates from the same person. Each colored branch and isolate label represents a different outbreak.
represented 100% bootstrap support. Bootstrap support was estimated only for kSNP and cgMLST ML phylogenetic trees; the Bayesian recombination-adjusted phylogenies from ClonalFrameML do not estimate bootstrap values. Isolate label contains isolate ID, CC, PFGE pattern, outbreak or sporadic isolates, and year. Geographically matched sporadic and outbreak isolates are indicated by the same number following “SP” or “OB” in label. M26263 and M26417 were isolates from the same person. Each colored branch and isolate label represents a different outbreak.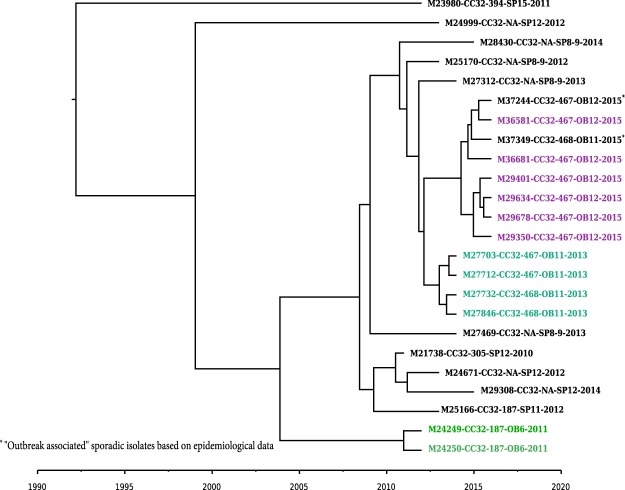
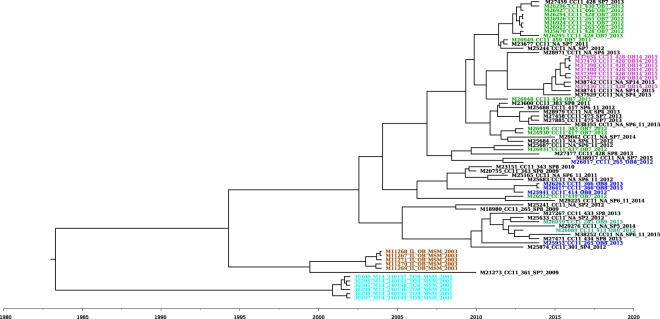
References
-
- Cohn AC, et al. Prevention and control of meningococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP) MMWR Recomm. Rep. 2013;62:1–28. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
