

Artificial Intelligence Approach for Variant Reporting
- PMID: 30364844
- PMCID: PMC6198661
- DOI: 10.1200/CCI.16.00079
Artificial Intelligence Approach for Variant Reporting
Abstract
Purpose: Next-generation sequencing technologies are actively applied in clinical oncology. Bioinformatics pipeline analysis is an integral part of this process; however, humans cannot yet realize the full potential of the highly complex pipeline output. As a result, the decision to include a variant in the final report during routine clinical sign-out remains challenging.
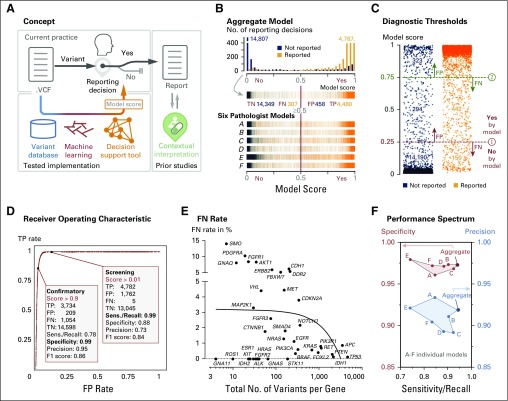
Methods: We used an artificial intelligence approach to capture the collective clinical sign-out experience of six board-certified molecular pathologists to build and validate a decision support tool for variant reporting. We extracted all reviewed and reported variants from our clinical database and tested several machine learning models. We used 10-fold cross-validation for our variant call prediction model, which derives a contiguous prediction score from 0 to 1 (no to yes) for clinical reporting.
Results: For each of the 19,594 initial training variants, our pipeline generates approximately 500 features, which results in a matrix of > 9 million data points. From a comparison of naive Bayes, decision trees, random forests, and logistic regression models, we selected models that allow human interpretability of the prediction score. The logistic regression model demonstrated 1% false negativity and 2% false positivity. The final models' Youden indices were 0.87 and 0.77 for screening and confirmatory cutoffs, respectively. Retraining on a new assay and performance assessment in 16,123 independent variants validated our approach (Youden index, 0.93). We also derived individual pathologist-centric models (virtual consensus conference function), and a visual drill-down functionality allows assessment of how underlying features contributed to a particular score or decision branch for clinical implementation.
Conclusion: Our decision support tool for variant reporting is a practically relevant artificial intelligence approach to harness the next-generation sequencing bioinformatics pipeline output when the complexity of data interpretation exceeds human capabilities.
Conflict of interest statement
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc. Michael G. Zomnir No relationship to disclose Lev Lipkin Stock and Other Ownership Interests: TEVA Pharmaceuticals Industries, Pfizer, Novartis Maciej Pacula Patents, Royalties, Other Intellectual Property: Ute Geigenmuller, Doris Damian, Maciej Pacula, Mark A. DePristo. Methods and Systems for Determining Autism Spectrum Disorder Risk (US patent 9,176,113), granted November 3, 2015 (Inst) Enrique Dominguez Meneses No relationship to disclose Allison MacLeay Travel, Accommodations, Expenses: InterSystems, Athenahealth (I) Sekhar Duraisamy No relationship to disclose Nishchal Nadhamuni No relationship to disclose
Figures


References
-
- Haber DA, Gray NS, Baselga J: The evolving war on cancer. Cell 145:19-24, 2011 - PubMed
-
- Buermans HP, den Dunnen JT: Next generation sequencing technology: Advances and applications. Biochim Biophys Acta 1842:1932-1941, 2014 - PubMed
-
- Hagemann IS, O’Neill PK, Erill I, et al. : Diagnostic yield of targeted next generation sequencing in various cancer types: An information-theoretic approach. Cancer Genet 208:441-447, 2015 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
