

Expression and epigenetic regulatory mechanism of BNIP3 in clear cell renal cell carcinoma
- PMID: 30365137
- PMCID: PMC6254932
- DOI: 10.3892/ijo.2018.4603
Expression and epigenetic regulatory mechanism of BNIP3 in clear cell renal cell carcinoma
Abstract
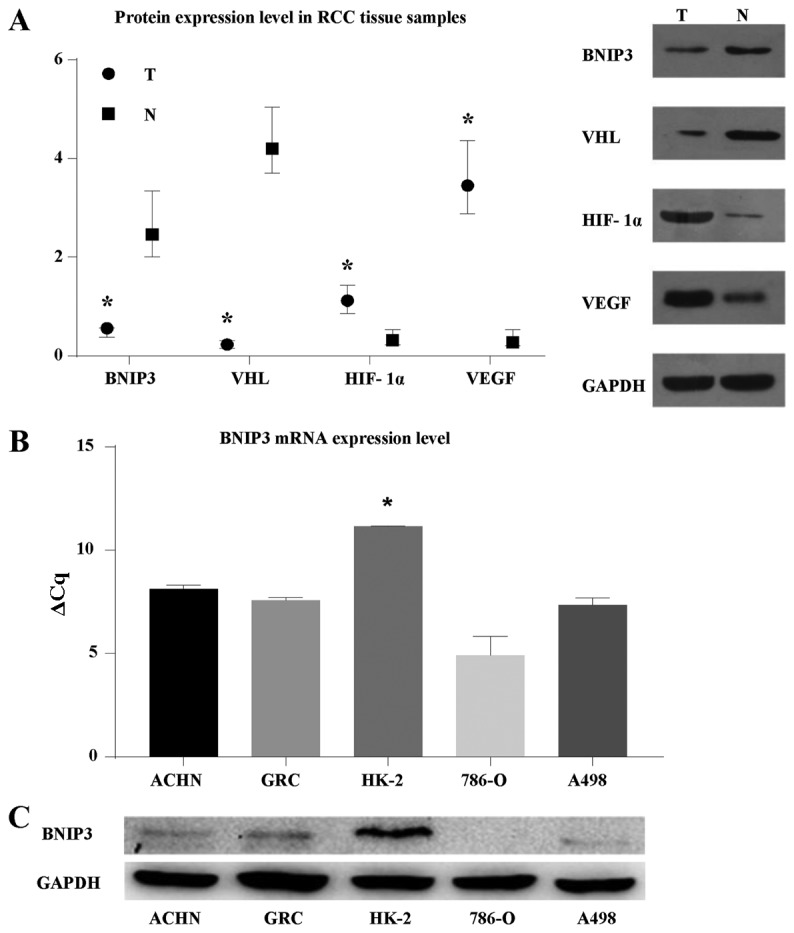
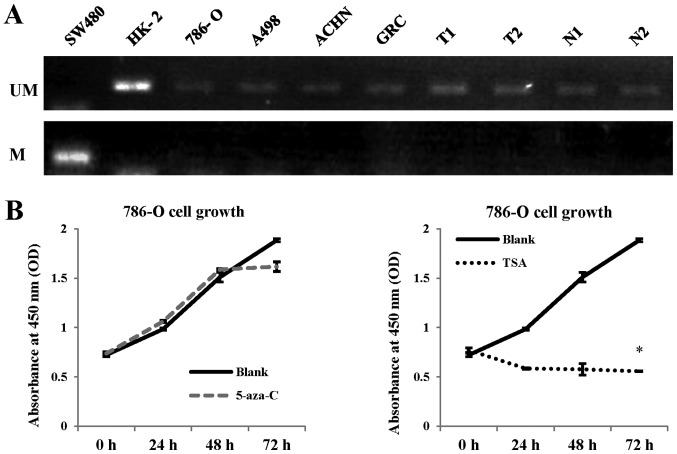
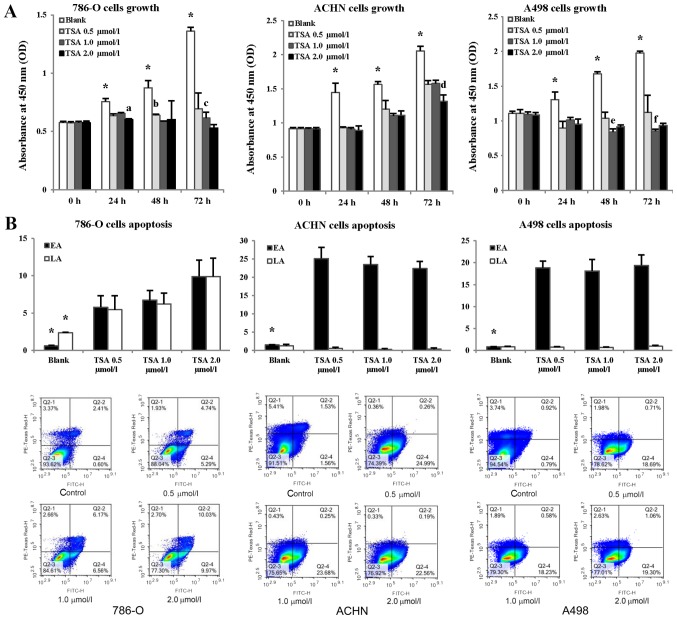
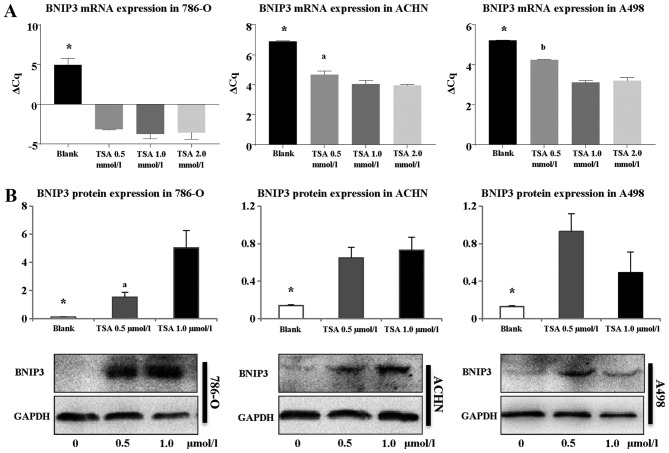
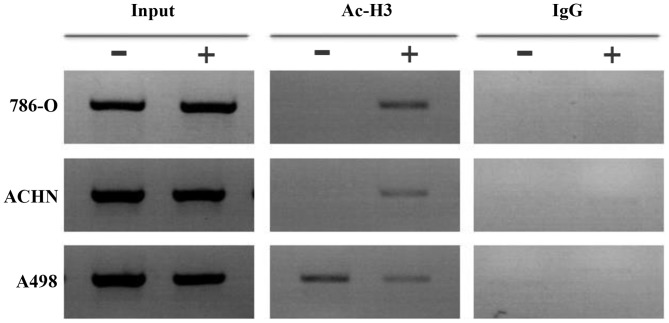
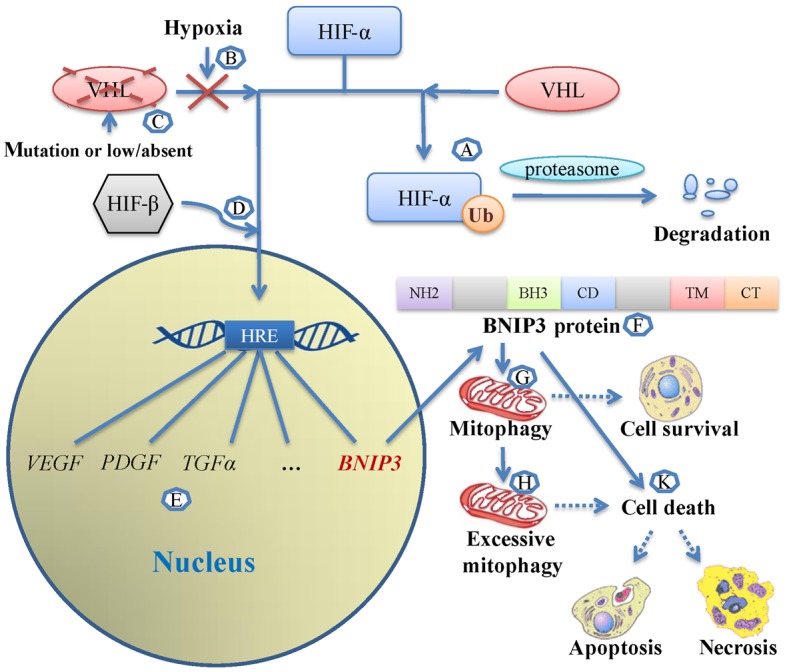
The majority of clear cell renal cell carcinomas (ccRCCs) are caused by an accumulation of hypoxia‑inducible factor (HIF) and the overexpression of downstream genes in response to the von Hippel‑Lindau (VHL) gene becoming inactivated. In the present study, our hypothesis was that BNIP3, a gene positioned downstream of HIF, would be expressed at a higher level in ccRCC; however, instead, lower levels of BNIP3 expression were identified in RCC tumor tissues compared with adjacent non‑tumor tissues. These changes were associated with lower levels of VHL, and higher levels of HIF and vascular endothelial growth factor. BNIP3 was also undetectable in three investigated RCC cell lines (786‑O, ACHN, A498) and GRC‑1‑1 cells. Methylation of the BNIP3 promoter was not detected, and neither did treatment with a methylation inhibitor cause cell proliferation. However, treatment with a histone deacetylation inhibitor, trichostatin A (TSA), inhibited cultured RCC cell proliferation, promoted apoptosis and restored BNIP3 expression. Furthermore, histone deacetylation of the BNIP3 promoter was identified in ACHN and 786‑O cells, and the acetylation status was restored following TSA treatment. Taken together, the results of the present study suggest that histone deacetylation, but not methylation, is most likely to cause BNIP3 inactivation in RCC. The data also indicated that restoration of BNIP3 expression by a histone deacetylation inhibitor led to growth inhibition and apoptotic promotion in RCC.
Keywords: carcinoma; renal cell; hypoxia-inducible factor; Bcl-2/adenovirus E1B 19 kDa interacting protein 3; BNIP3; DNA methylation; histone deacetylation; epigenetic regulation.
Figures
References
-
- Bultitude MF. Campbell-Walsh Urology. 10th edition. BJU International; New York, NY: 2012.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
