

Pharmacological targeting of RAS: Recent success with direct inhibitors
- PMID: 30366101
- PMCID: PMC6360110
- DOI: 10.1016/j.phrs.2018.10.021
Pharmacological targeting of RAS: Recent success with direct inhibitors
Abstract
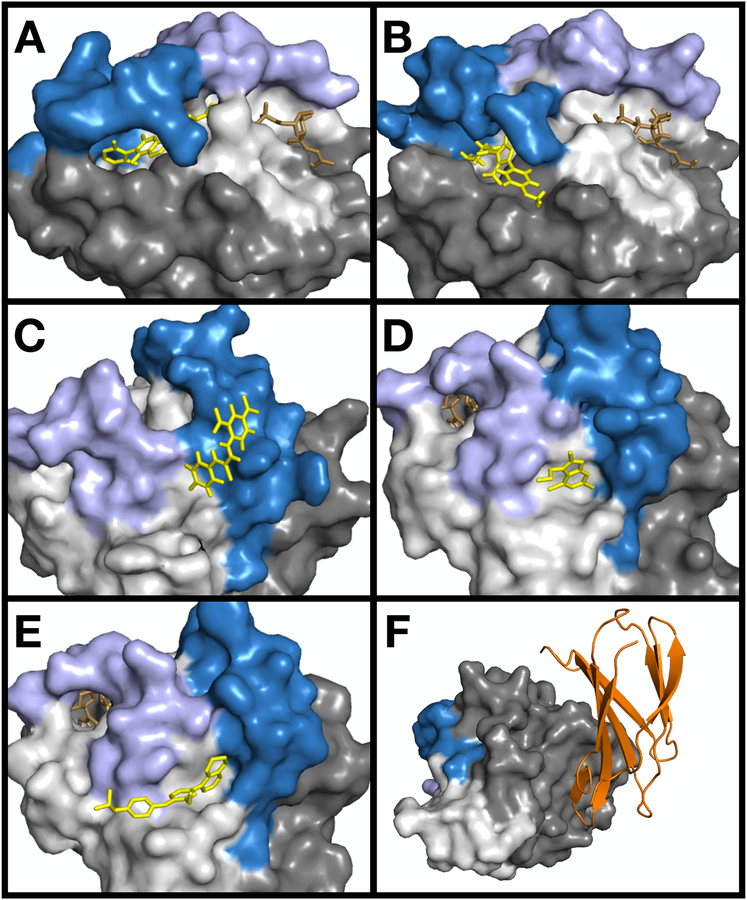
RAS has long been viewed as undruggable due to its lack of deep pockets for binding of small molecule inhibitors. However, recent successes in the development of direct RAS inhibitors suggest that the goal of pharmacological inhibition of RAS in patients may soon be realized. This review will discuss the role of RAS in cancer, the approaches used to develop direct RAS inhibitors, and highlight recent successes in the development of novel RAS inhibitory compounds that target different aspects of RAS biochemistry. In particular, this review will discuss the different properties of RAS that have been targeted by various inhibitors including membrane localization, the different activation states of RAS, effector binding, and nucleotide exchange. In addition, this review will highlight the recent success with mutation-specific inhibitors that exploit the unique biochemistry of the RAS(G12C) mutant. Although this mutation in KRAS accounts for 11% of all KRAS mutations in cancer, it is the most prominent KRAS mutant in lung cancer suggesting that G12C-specific inhibitors may provide a new approach for treating the subset of lung cancer patients harboring this mutant allele. Finally, this review will discuss the involvement of dimerization in RAS function and highlight new approaches to inhibit RAS by specifically interfering with RAS:RAS interaction.
Keywords: 3144 (PubChem CID 102004330); ABD7 (PubChem CID 134812710); ARS-1620 (PubChem CID 132274053); BIM-46187 (PubChem CID 11593027); CAAX motif; Cancer; DCAI, PubChem CID 1381961; Deltarasin (PubChem CID 73292904); Effector interaction; GTPase; IND12 (PubChem CID 76715657); Kobe2601 (PubChem CID 163309612); Monobody; Nucleotide exchange; RAS inhibitor; Sulindac sulfide (PubChem CID 5352624); Zn-cyclen (PubChem CID 129651749).
Published by Elsevier Ltd.
Conflict of interest statement
Conflict of Interest Statement:
The author declares that he has no conflicts of interest.
Figures



References
-
- Brummelkamp TR, Bernards R, Agami R, Stable suppression of tumorigenicity by virus-mediated RNA interference, Cancer Cell 2(3) (2002) 243–7. - PubMed
-
- Lim KH, Counter CM, Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance, Cancer Cell 8(5) (2005) 381–92. - PubMed
-
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, et al. , Essential role for oncogenic Ras in tumour maintenance, Nature (London) 400(6743) (1999) 468–72. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
