

Mutant TRP53 exerts a target gene-selective dominant-negative effect to drive tumor development
- PMID: 30366906
- PMCID: PMC6217734
- DOI: 10.1101/gad.314286.118
Mutant TRP53 exerts a target gene-selective dominant-negative effect to drive tumor development
Abstract
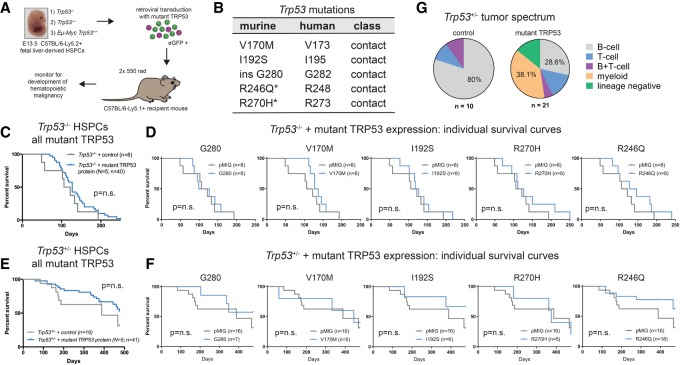
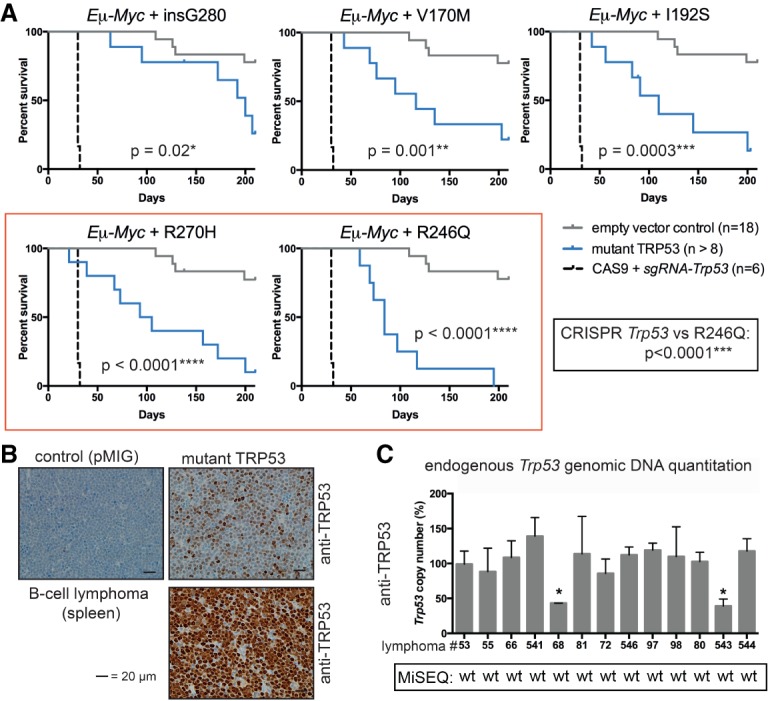
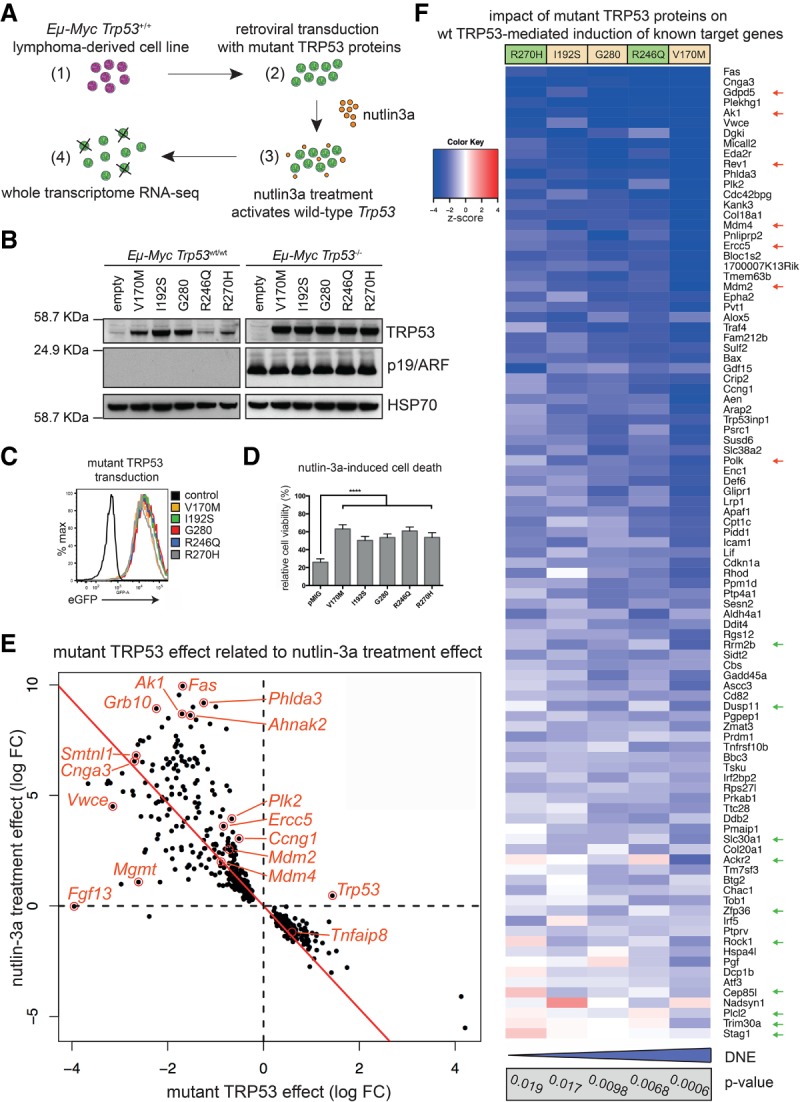
Mutations in Trp53, prevalent in human cancer, are reported to drive tumorigenesis through dominant-negative effects (DNEs) over wild-type TRP53 function as well as neomorphic gain-of-function (GOF) activity. We show that five TRP53 mutants do not accelerate lymphomagenesis on a TRP53-deficient background but strongly synergize with c-MYC overexpression in a manner that distinguishes the hot spot Trp53 mutations. RNA sequencing revealed that the mutant TRP53 DNE does not globally repress wild-type TRP53 function but disproportionately impacts a subset of wild-type TRP53 target genes. Accordingly, TRP53 mutant proteins impair pathways for DNA repair, proliferation, and metabolism in premalignant cells. This reveals that, in our studies of lymphomagenesis, mutant TRP53 drives tumorigenesis primarily through the DNE, which modulates wild-type TRP53 function in a manner advantageous for neoplastic transformation.
Keywords: TRP53; TRP53 target genes; dominant-negative effect; tumorigenesis.
© 2018 Aubrey et al.; Published by Cold Spring Harbor Laboratory Press.
Figures

References
-
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. 1985. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318: 533–538. - PubMed
-
- Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O'Connor L, Milla L, Wilcox S, Tai L, Strasser A, Herold MJ. 2015. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep 10: 1422–1432. - PubMed
-
- Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, Olivier M. 2016. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat 37: 865–876. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials