

FAM210B is an erythropoietin target and regulates erythroid heme synthesis by controlling mitochondrial iron import and ferrochelatase activity
- PMID: 30366982
- PMCID: PMC6314115
- DOI: 10.1074/jbc.RA118.002742
FAM210B is an erythropoietin target and regulates erythroid heme synthesis by controlling mitochondrial iron import and ferrochelatase activity
Abstract
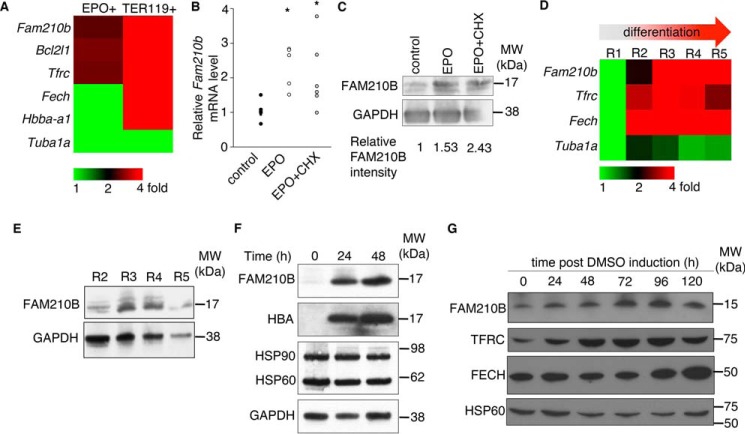
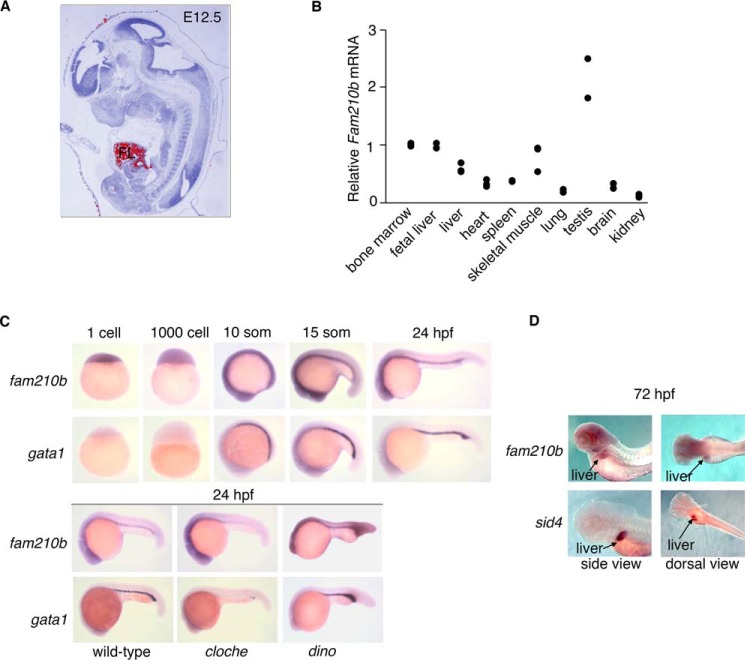
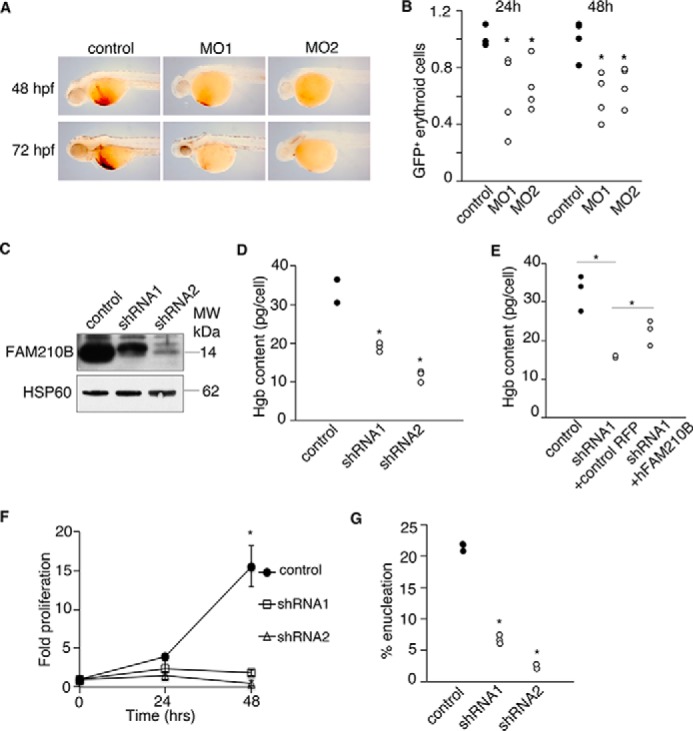
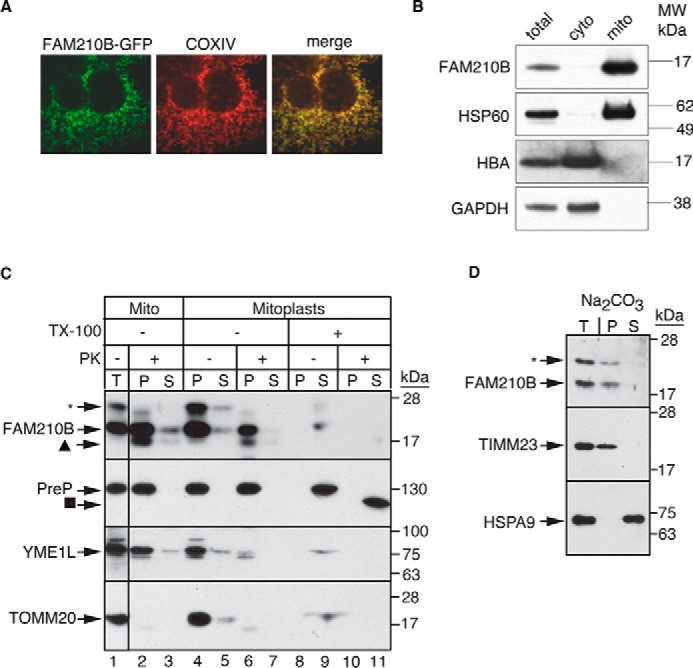
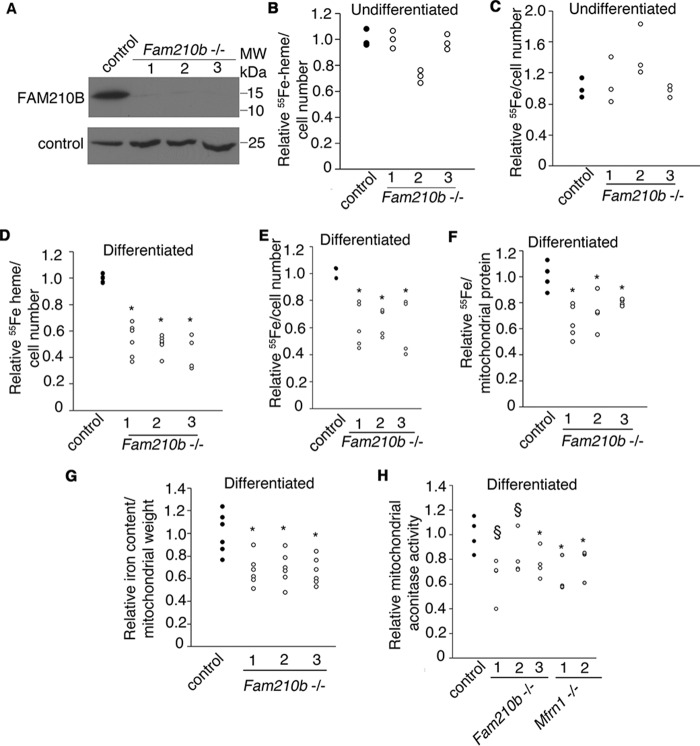
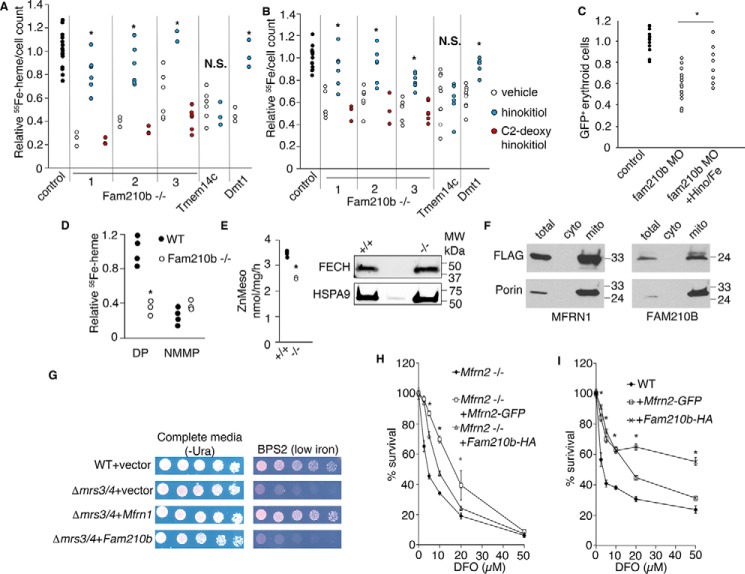
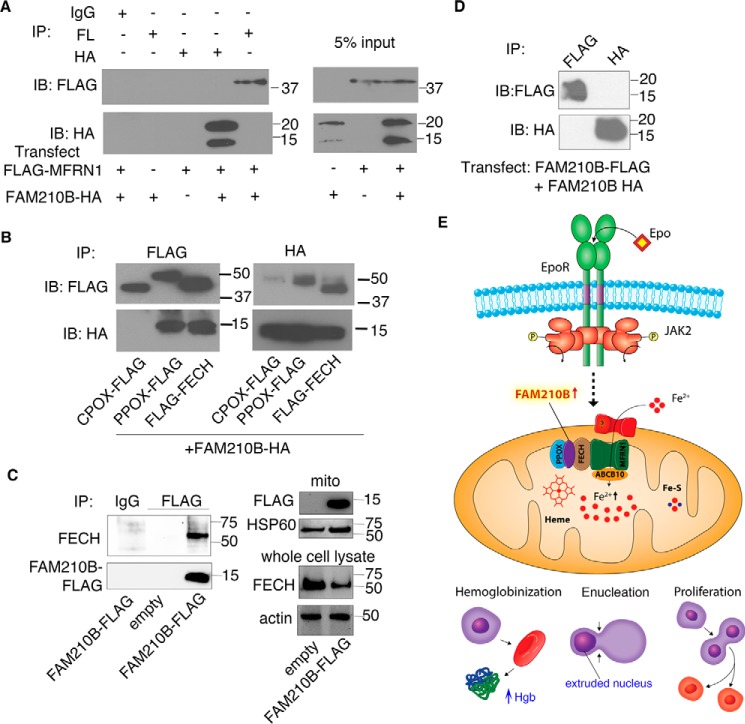
Erythropoietin (EPO) signaling is critical to many processes essential to terminal erythropoiesis. Despite the centrality of iron metabolism to erythropoiesis, the mechanisms by which EPO regulates iron status are not well-understood. To this end, here we profiled gene expression in EPO-treated 32D pro-B cells and developing fetal liver erythroid cells to identify additional iron regulatory genes. We determined that FAM210B, a mitochondrial inner-membrane protein, is essential for hemoglobinization, proliferation, and enucleation during terminal erythroid maturation. Fam210b deficiency led to defects in mitochondrial iron uptake, heme synthesis, and iron-sulfur cluster formation. These defects were corrected with a lipid-soluble, small-molecule iron transporter, hinokitiol, in Fam210b-deficient murine erythroid cells and zebrafish morphants. Genetic complementation experiments revealed that FAM210B is not a mitochondrial iron transporter but is required for adequate mitochondrial iron import to sustain heme synthesis and iron-sulfur cluster formation during erythroid differentiation. FAM210B was also required for maximal ferrochelatase activity in differentiating erythroid cells. We propose that FAM210B functions as an adaptor protein that facilitates the formation of an oligomeric mitochondrial iron transport complex, required for the increase in iron acquisition for heme synthesis during terminal erythropoiesis. Collectively, our results reveal a critical mechanism by which EPO signaling regulates terminal erythropoiesis and iron metabolism.
Keywords: cell metabolism; erythrocyte; erythropoiesis; heme; iron metabolism; red blood cell.
© 2018 Yien et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Gammella E., Diaz V., Recalcati S., Buratti P., Samaja M., Dey S., Noguchi C. T., Gassmann M., and Cairo G. (2015) Erythropoietin's inhibiting impact on hepcidin expression occurs indirectly. Am. J. Physiol. Regul. Integr. Comp. Physiol. 308, R330–R335 10.1152/ajpregu.00410.2014 - DOI - PMC - PubMed
-
- Nai A., Rubio A., Campanella A., Gourbeyre O., Artuso I., Bordini J., Gineste A., Latour C., Besson-Fournier C., Lin H. Y., Coppin H., Roth M.-P., Camaschella C., Silvestri L., Meynard D., Franke K., et al. (2016) Limiting hepatic Bmp-Smad signaling by matriptase-2 is required for erythropoietin-mediated hepcidin suppression in mice. Blood 127, 99–101 10.1182/blood-2015-11-681494 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL116364/HL/NHLBI NIH HHS/United States
- R01 DK030534/DK/NIDDK NIH HHS/United States
- P30 DK034854/DK/NIDDK NIH HHS/United States
- U54 DK110805/DK/NIDDK NIH HHS/United States
- U54 DK110858/DK/NIDDK NIH HHS/United States
- U54 DK083909/DK/NIDDK NIH HHS/United States
- R01 DK020503/DK/NIDDK NIH HHS/United States
- F32 DK098866/DK/NIDDK NIH HHS/United States
- K08 DK093705/DK/NIDDK NIH HHS/United States
- R24 OD017870/OD/NIH HHS/United States
- R35 GM118185/GM/NIGMS NIH HHS/United States
- R03 DK118307/DK/NIDDK NIH HHS/United States
- R01 GM061721/GM/NIGMS NIH HHS/United States
- P01 CA163222/CA/NCI NIH HHS/United States
- K01 DK106156/DK/NIDDK NIH HHS/United States
- R01 DK071116/DK/NIDDK NIH HHS/United States
- R01 GM073981/GM/NIGMS NIH HHS/United States
- P01 HL032262/HL/NHLBI NIH HHS/United States
- R37 DK030534/DK/NIDDK NIH HHS/United States
- R01 DK070838/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
