

Optimization of carbon and energy utilization through differential translational efficiency
- PMID: 30367068
- PMCID: PMC6203783
- DOI: 10.1038/s41467-018-06993-6
Optimization of carbon and energy utilization through differential translational efficiency
Abstract
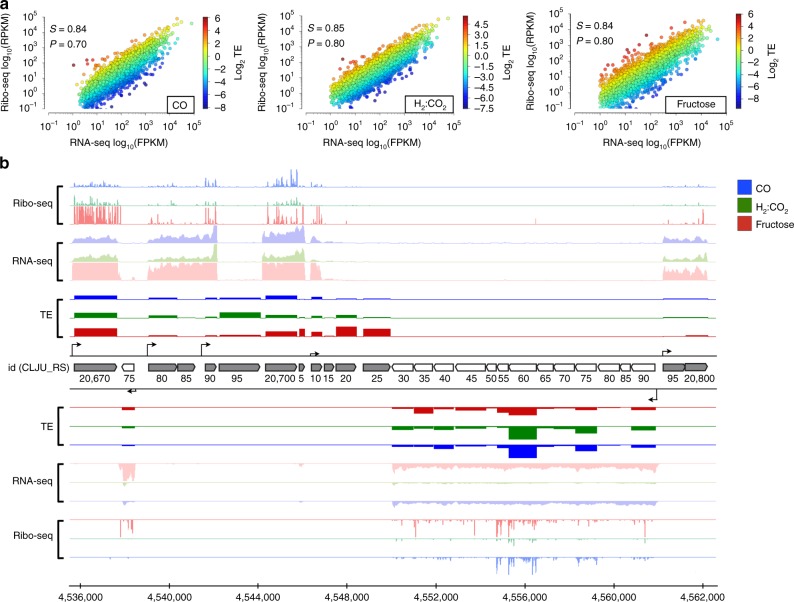
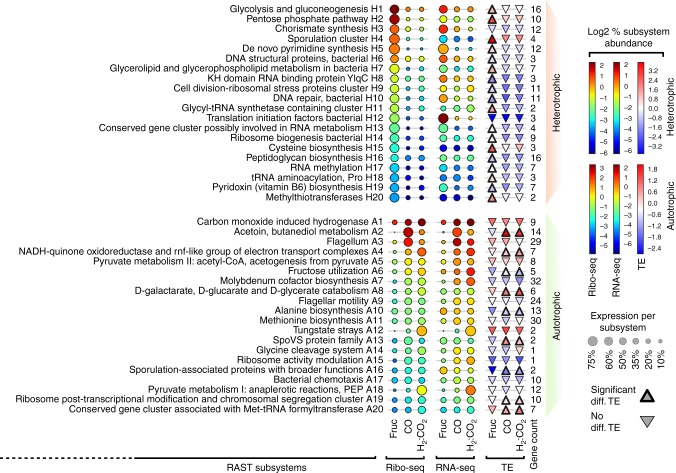
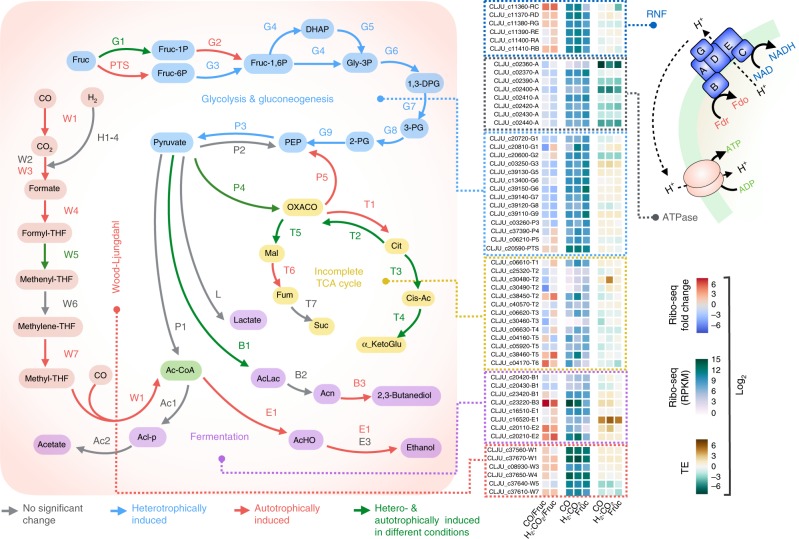
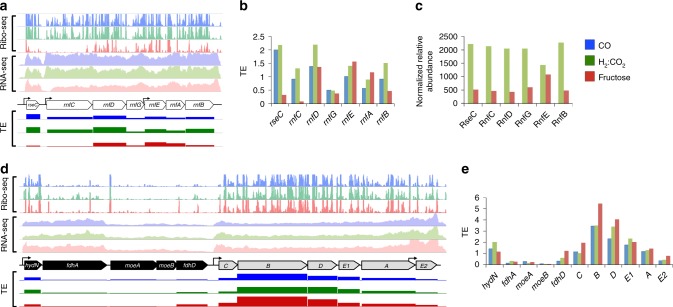
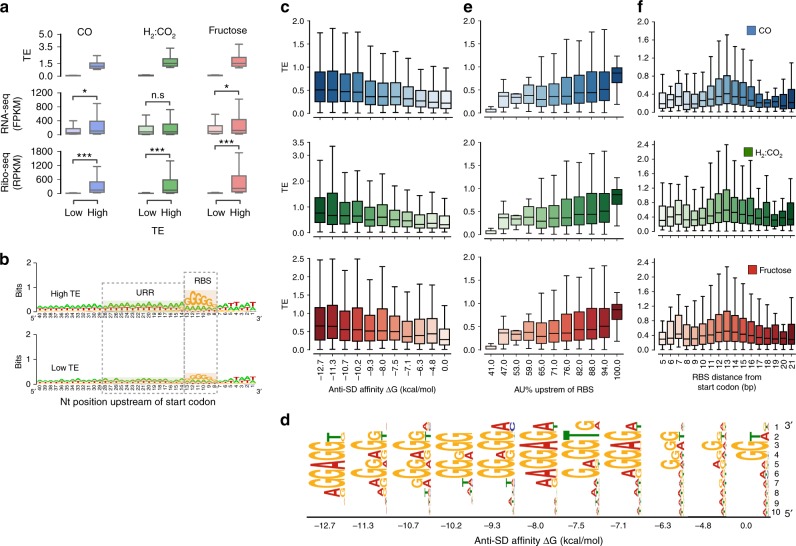
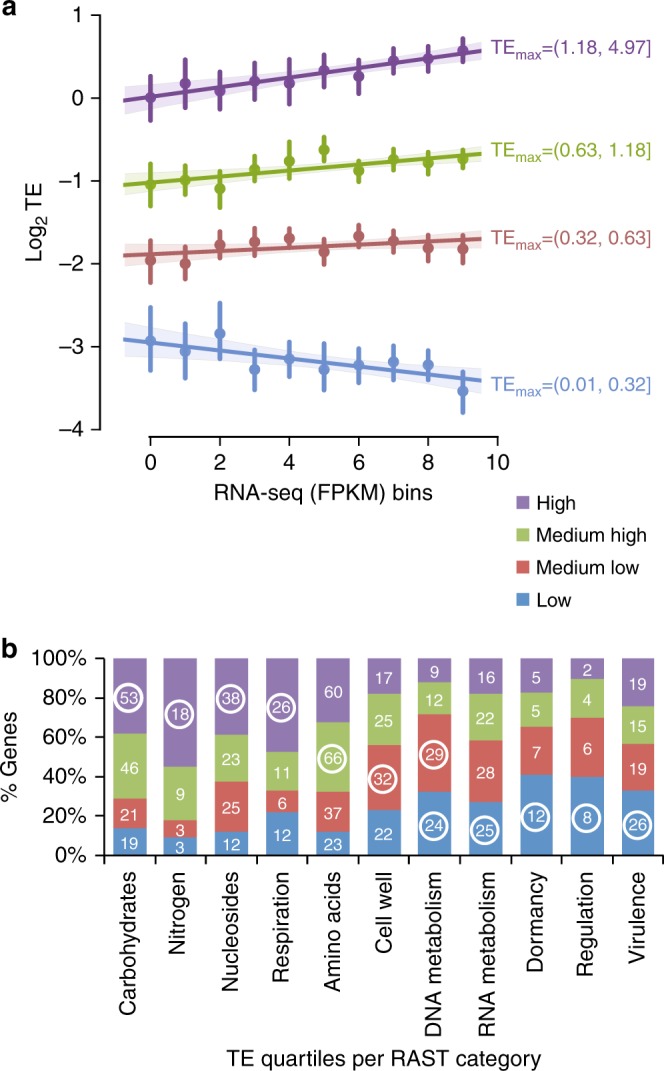
Control of translation is vital to all species. Here we employ a multi-omics approach to decipher condition-dependent translational regulation in the model acetogen Clostridium ljungdahlii. Integration of data from cells grown autotrophically or heterotrophically revealed that pathways critical to carbon and energy metabolism are under strong translational regulation. Major pathways involved in carbon and energy metabolism are not only differentially transcribed and translated, but their translational efficiencies are differentially elevated in response to resource availability under different growth conditions. We show that translational efficiency is not static and that it changes dynamically in response to mRNA expression levels. mRNAs harboring optimized 5'-untranslated region and coding region features, have higher translational efficiencies and are significantly enriched in genes encoding carbon and energy metabolism. In contrast, mRNAs enriched in housekeeping functions harbor sub-optimal features and have lower translational efficiencies. We propose that regulation of translational efficiency is crucial for effectively controlling resource allocation in energy-deprived microorganisms.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
