

A graph-based approach for proteoform identification and quantification using top-down homogeneous multiplexed tandem mass spectra
- PMID: 30367573
- PMCID: PMC6101081
- DOI: 10.1186/s12859-018-2273-4
A graph-based approach for proteoform identification and quantification using top-down homogeneous multiplexed tandem mass spectra
Abstract
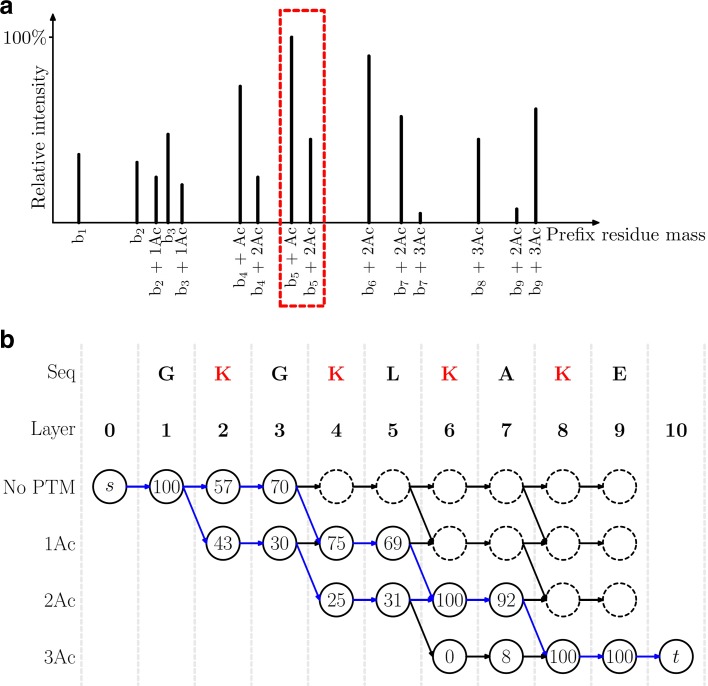
Background: Top-down homogeneous multiplexed tandem mass (HomMTM) spectra are generated from modified proteoforms of the same protein with different post-translational modification patterns. They are frequently observed in the analysis of ultramodified proteins, some proteoforms of which have similar molecular weights and cannot be well separated by liquid chromatography in mass spectrometry analysis.
Results: We formulate the top-down HomMTM spectral identification problem as the minimum error k-splittable flow problem on graphs and propose a graph-based algorithm for the identification and quantification of proteoforms using top-down HomMTM spectra.
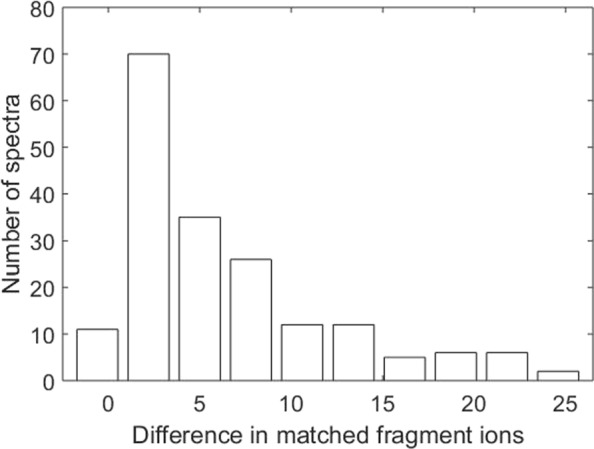
Conclusions: Experiments on a top-down mass spectrometry data set of the histone H4 protein showed that the proposed method identified many proteoform pairs that better explain the query spectra than single proteoforms.
Keywords: Graph algorithms; Mass spectrometry; Multiplexed mass spectra; Top-down.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- DiMaggio Jr PA, Young NL, Baliban RC, Garcia BA, Floudas CA. A mixed integer linear optimization framework for the identification and quantification of targeted post-translational modifications of highly modified proteins using multiplexed electron transfer dissociation tandem mass spectrometry. Mol Cell Proteomics. 2009;8:2527–43. doi: 10.1074/mcp.M900144-MCP200. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
