

CRISPR -Mediated Expression of the Fetal Scn5a Isoform in Adult Mice Causes Conduction Defects and Arrhythmias
- PMID: 30371314
- PMCID: PMC6404881
- DOI: 10.1161/JAHA.118.010393
CRISPR -Mediated Expression of the Fetal Scn5a Isoform in Adult Mice Causes Conduction Defects and Arrhythmias
Abstract
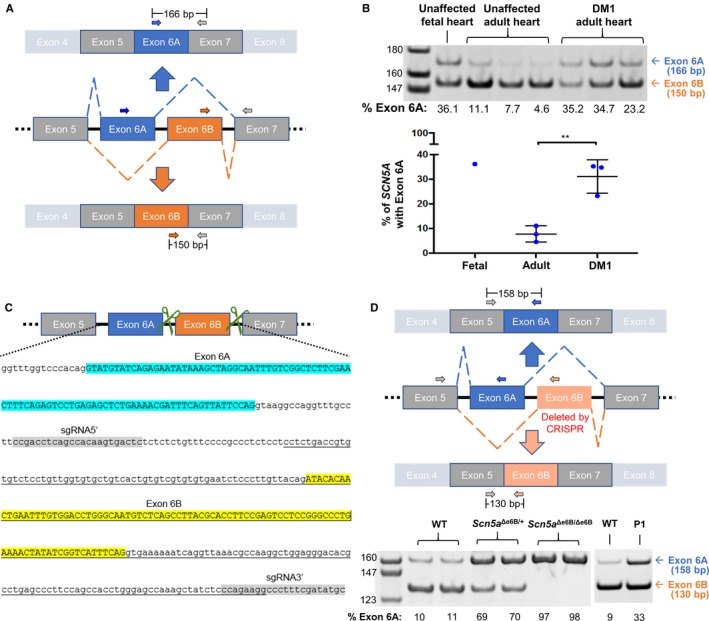
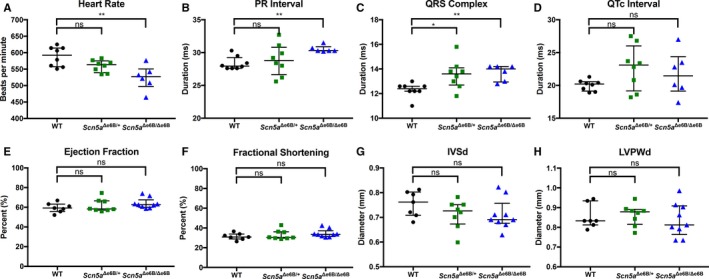
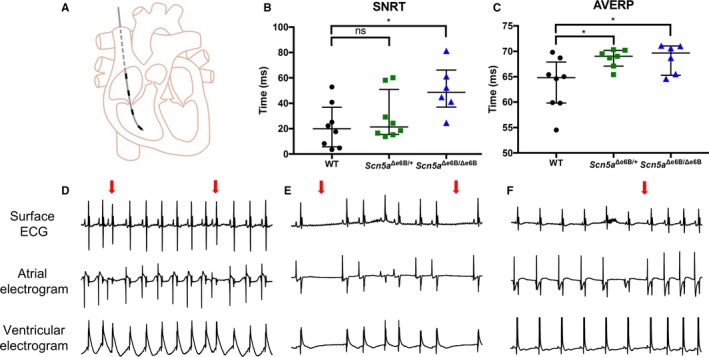
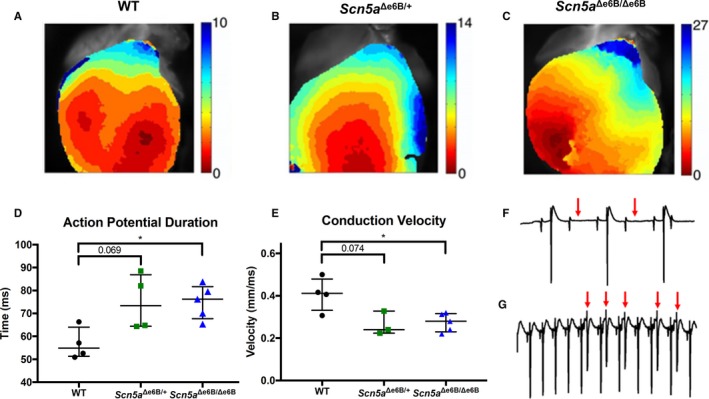
Background The sodium channel, Nav1.5, encoded by SCN 5A, undergoes developmentally regulated splicing from inclusion of exon 6A in the fetal heart to exon 6B in adults. These mutually exclusive exons differ in 7 amino acids altering the electrophysiological properties of the Nav1.5 channel. In myotonic dystrophy type 1, SCN 5A is mis-spliced such that the fetal pattern of exon 6A inclusion is detected in adult hearts. Cardiac manifestations of myotonic dystrophy type 1 include conduction defects and arrhythmias and are the second-leading cause of death. Methods and Results This work aimed to determine the impact of SCN 5A mis-splicing on cardiac function. We used clustered regularly interspaced short palindromic repeat ( CRISPR) /CRISPR-associated protein 9 (Cas9) to delete Scn5a exon 6B in mice, thereby redirecting splicing toward exon 6A. These mice exhibit prolonged PR and QRS intervals, slowed conduction velocity, extended action potential duration, and are highly susceptible to arrhythmias. Conclusions Our findings highlight a nonmutational pathological mechanism of arrhythmias and conduction defects as a result of mis-splicing of the predominant cardiac sodium channel. Animals homozygous for the deleted exon express only the fetal isoform and have more-severe phenotypes than heterozygotes that also express the adult isoform. This observation is directly relevant to myotonic dystrophy type 1, and possibly pathological arrhythmias, in which individuals differ with regard to the ratios of the isoforms expressed.
Keywords: SCN5A; alternative splicing; arrhythmia; conduction; myotonic dystrophy cardiomyopathy.
Figures
Similar articles
-
Rescue of Scn5a mis-splicing does not improve the structural and functional heart defects of a DM1 heart mouse model.Hum Mol Genet. 2024 Oct 7;33(20):1789-1799. doi: 10.1093/hmg/ddae117. Hum Mol Genet. 2024. PMID: 39126705 Free PMC article.
-
Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy.Nat Commun. 2016 Apr 11;7:11067. doi: 10.1038/ncomms11067. Nat Commun. 2016. PMID: 27063795 Free PMC article.
-
Maturation of hiPSC-derived cardiomyocytes promotes adult alternative splicing of SCN5A and reveals changes in sodium current associated with cardiac arrhythmia.Cardiovasc Res. 2023 Mar 17;119(1):167-182. doi: 10.1093/cvr/cvac059. Cardiovasc Res. 2023. PMID: 35394010 Free PMC article.
-
[Myotonia and cardiac conduction defects in myotonic dystrophy and defect in ion channels].Rinsho Byori. 2014 Mar;62(3):246-54. Rinsho Byori. 2014. PMID: 24800500 Review. Japanese.
-
Arrhythmic Phenotypes Are a Defining Feature of Dilated Cardiomyopathy-Associated SCN5A Variants: A Systematic Review.Circ Genom Precis Med. 2022 Feb;15(1):e003432. doi: 10.1161/CIRCGEN.121.003432. Epub 2021 Dec 24. Circ Genom Precis Med. 2022. PMID: 34949099
Cited by
-
Clinical and Molecular Insights into Gastrointestinal Dysfunction in Myotonic Dystrophy Types 1 & 2.Int J Mol Sci. 2022 Nov 26;23(23):14779. doi: 10.3390/ijms232314779. Int J Mol Sci. 2022. PMID: 36499107 Free PMC article. Review.
-
Mice lacking MBNL1 and MBNL2 exhibit sudden cardiac death and molecular signatures recapitulating myotonic dystrophy.Hum Mol Genet. 2022 Sep 10;31(18):3144-3160. doi: 10.1093/hmg/ddac108. Hum Mol Genet. 2022. PMID: 35567413 Free PMC article.
-
iPSC-derived cardiomyocytes from patients with myotonic dystrophy type 1 have abnormal ion channel functions and slower conduction velocities.Sci Rep. 2021 Jan 28;11(1):2500. doi: 10.1038/s41598-021-82007-8. Sci Rep. 2021. PMID: 33510259 Free PMC article.
-
Development and disease-specific regulation of RNA splicing in cardiovascular system.Front Cell Dev Biol. 2024 Jul 9;12:1423553. doi: 10.3389/fcell.2024.1423553. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 39045460 Free PMC article. Review.
-
BlockmiR AONs as Site-Specific Therapeutic MBNL Modulation in Myotonic Dystrophy 2D and 3D Muscle Cells and HSALR Mice.Pharmaceutics. 2023 Mar 31;15(4):1118. doi: 10.3390/pharmaceutics15041118. Pharmaceutics. 2023. PMID: 37111604 Free PMC article.
References
-
- Klaver EC, Versluijs GM, Wilders R. Cardiac ion channel mutations in the sudden infant death syndrome. Int J Cardiol. 2011;152:162–170. - PubMed
-
- Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol. 2009;6:337–348. - PubMed
-
- Wang Q, Shen J, Li Z, Timothy K, Vincent GM, Priori SG, Schwartz PJ, Keating MT. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet. 1995;4:1603–1607. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
