

Identification of spatially associated subpopulations by combining scRNAseq and sequential fluorescence in situ hybridization data
- PMID: 30371680
- PMCID: PMC6488461
- DOI: 10.1038/nbt.4260
Identification of spatially associated subpopulations by combining scRNAseq and sequential fluorescence in situ hybridization data
Abstract
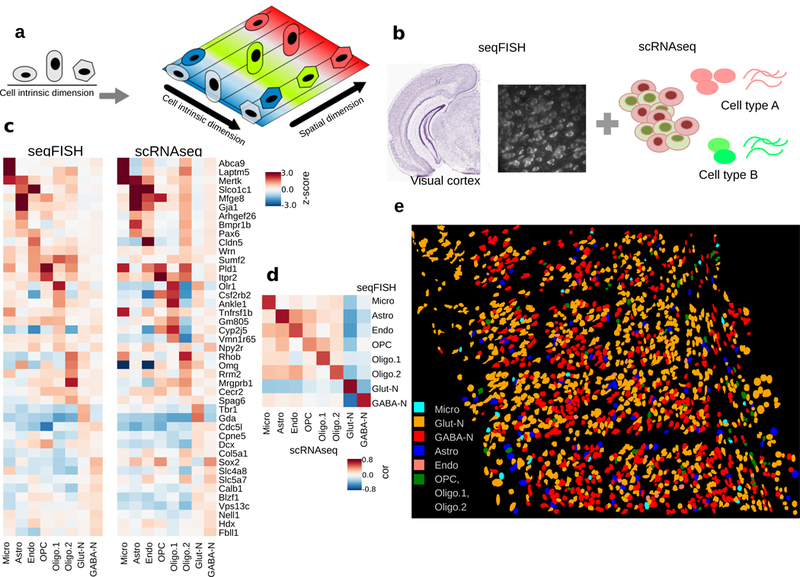
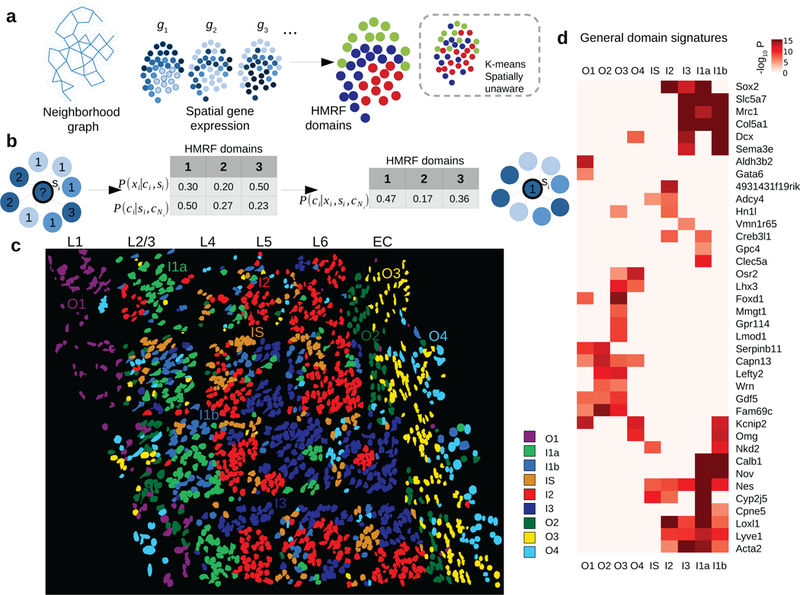
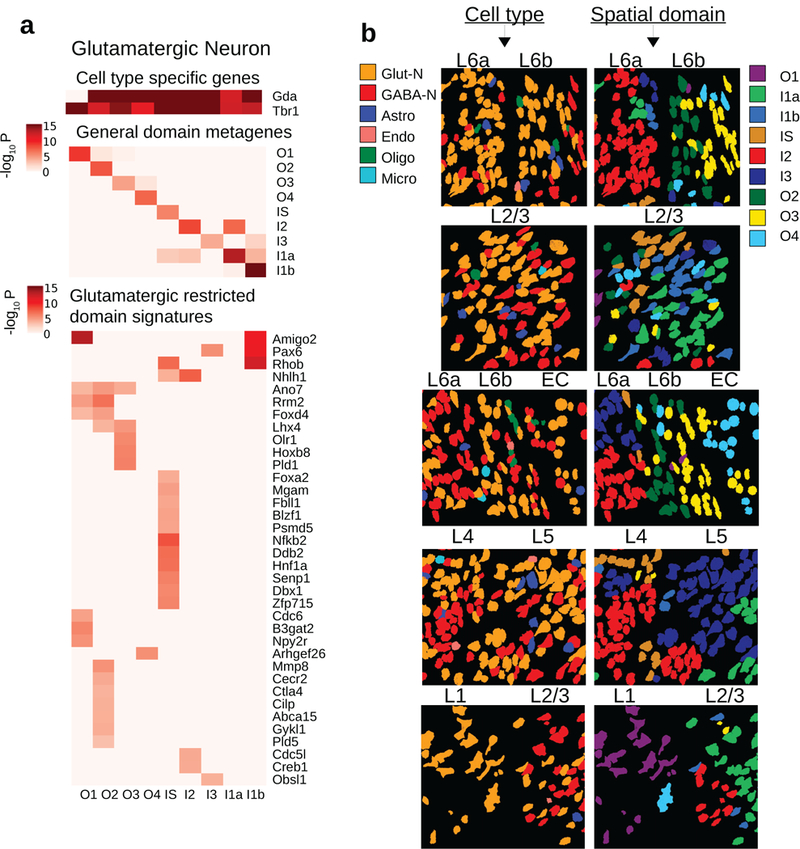
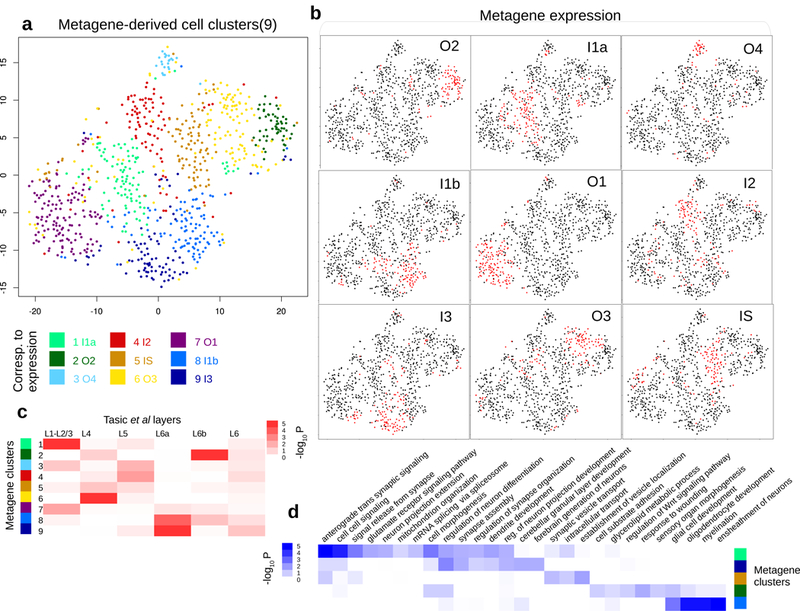
How intrinsic gene-regulatory networks interact with a cell's spatial environment to define its identity remains poorly understood. We developed an approach to distinguish between intrinsic and extrinsic effects on global gene expression by integrating analysis of sequencing-based and imaging-based single-cell transcriptomic profiles, using cross-platform cell type mapping combined with a hidden Markov random field model. We applied this approach to dissect the cell-type- and spatial-domain-associated heterogeneity in the mouse visual cortex region. Our analysis identified distinct spatially associated, cell-type-independent signatures in the glutamatergic and astrocyte cell compartments. Using these signatures to analyze single-cell RNA sequencing data, we identified previously unknown spatially associated subpopulations, which were validated by comparison with anatomical structures and Allen Brain Atlas images.
Conflict of interest statement
Competing Interests
None.
Figures
References
-
- Zhang J & Li L Stem cell niche: Microenvironment and beyond. J. Biol. Chem. 283, 9499–9503 (2008). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
