

Global variation in bacterial strains that cause tuberculosis disease: a systematic review and meta-analysis
- PMID: 30373589
- PMCID: PMC6206891
- DOI: 10.1186/s12916-018-1180-x
Global variation in bacterial strains that cause tuberculosis disease: a systematic review and meta-analysis
Abstract
Background: The host, microbial, and environmental factors that contribute to variation in tuberculosis (TB) disease are incompletely understood. Accumulating evidence suggests that one driver of geographic variation in TB disease is the local ecology of mycobacterial genotypes or strains, and there is a need for a comprehensive and systematic synthesis of these data. The objectives of this study were to (1) map the global distribution of genotypes that cause TB disease and (2) examine whether any epidemiologically relevant clinical characteristics were associated with those genotypes.
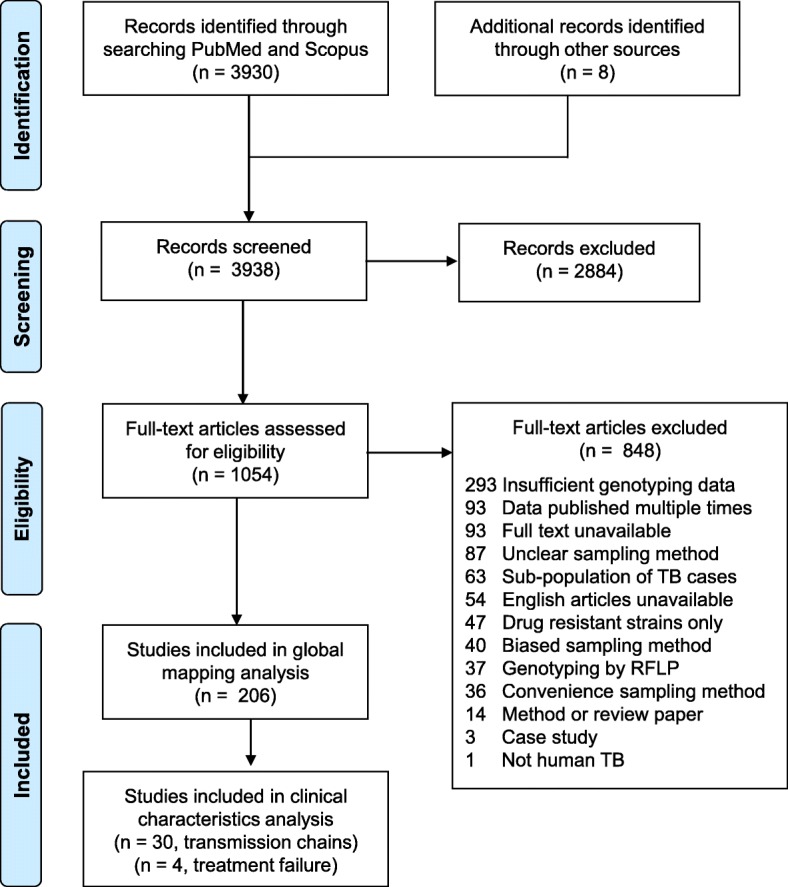
Methods: We performed a systematic review of PubMed and Scopus to create a comprehensive dataset of human TB molecular epidemiology studies that used representative sampling techniques. The methods were developed according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA). We extracted and synthesized data from studies that reported prevalence of bacterial genotypes and from studies that reported clinical characteristics associated with those genotypes.
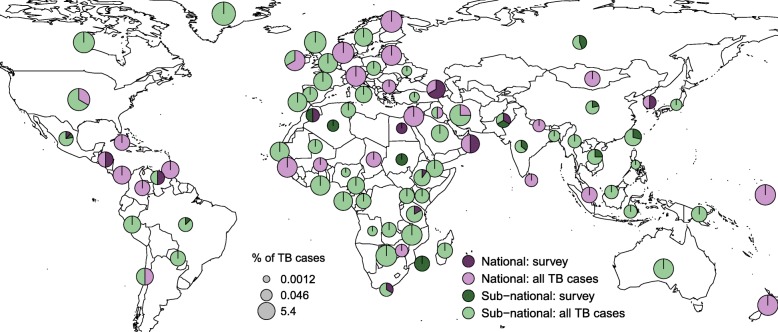
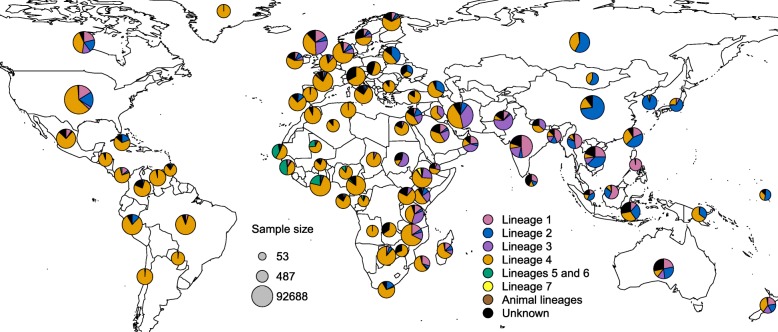
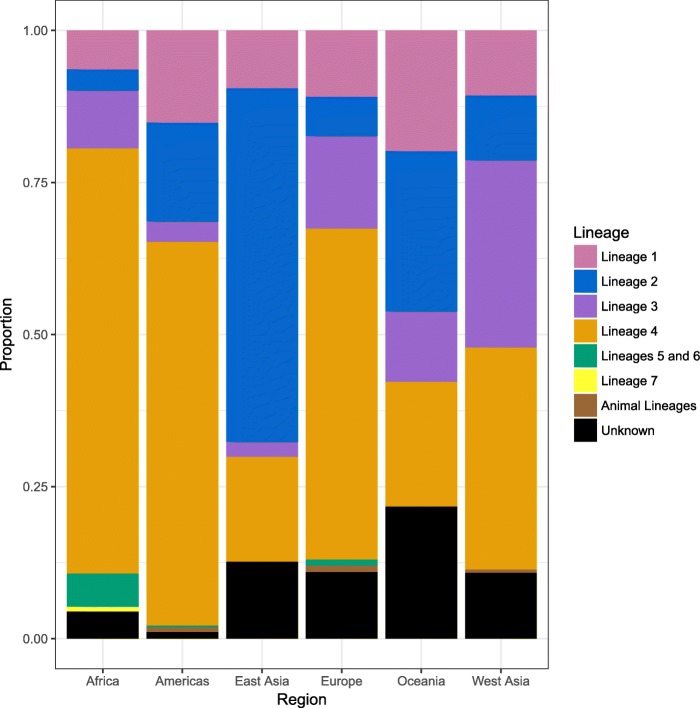
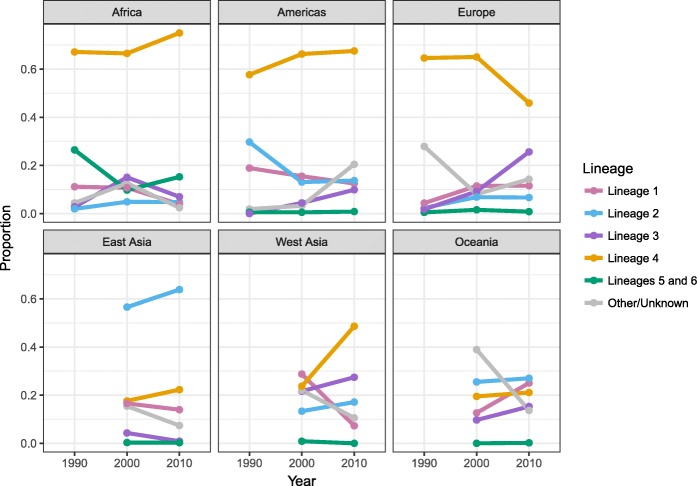
Results: The results of this study are twofold. First, we identified 206 studies for inclusion in the study, representing over 200,000 bacterial isolates collected over 27 years in 85 countries. We mapped the genotypes and found that, consistent with previously published maps, Euro-American lineage 4 and East Asian lineage 2 strains are widespread, and West African lineages 5 and 6 strains are geographically restricted. Second, 30 studies also reported transmission chains and 4 reported treatment failure associated with genotypes. We performed a meta-analysis and found substantial heterogeneity across studies. However, based on the data available, we found that lineage 2 strains may be associated with increased risk of transmission chains, while lineages 5 and 6 strains may be associated with reduced risk, compared with lineage 4 strains.
Conclusions: This study provides the most comprehensive systematic analysis of the evidence for diversity in bacterial strains that cause TB disease. The results show both geographic and epidemiological differences between strains, which could inform our understanding of the global burden of TB. Our findings also highlight the challenges of collecting the clinical data required to inform TB diagnosis and treatment. We urge future national TB programs and research efforts to prioritize and reinforce clinical data collection in study designs and results dissemination.
Keywords: Epidemiology; Genetic variation; Genotype; Molecular epidemiology; Mycobacterium tuberculosis; Tuberculosis.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Getahun H, Matteelli A, Chaisson RE, Raviglione M. Latent Mycobacterium tuberculosis infection. 10.1056/NEJMra1405427. 2015. doi:10.1056/NEJMra1405427. - PubMed
-
- Dye Christopher. The Population Biology of Tuberculosis. 2017.
-
- Abel Laurent, Fellay Jacques, Haas David W, Schurr Erwin, Srikrishna Geetha, Urbanowski Michael, Chaturvedi Nimisha, Srinivasan Sudha, Johnson Daniel H, Bishai William R. Genetics of human susceptibility to active and latent tuberculosis: present knowledge and future perspectives. The Lancet Infectious Diseases. 2018;18(3):e64–e75. doi: 10.1016/S1473-3099(17)30623-0. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
