

Arrhythmogenic Mechanisms in Heart Failure: Linking β-Adrenergic Stimulation, Stretch, and Calcium
- PMID: 30374311
- PMCID: PMC6196916
- DOI: 10.3389/fphys.2018.01453
Arrhythmogenic Mechanisms in Heart Failure: Linking β-Adrenergic Stimulation, Stretch, and Calcium
Abstract
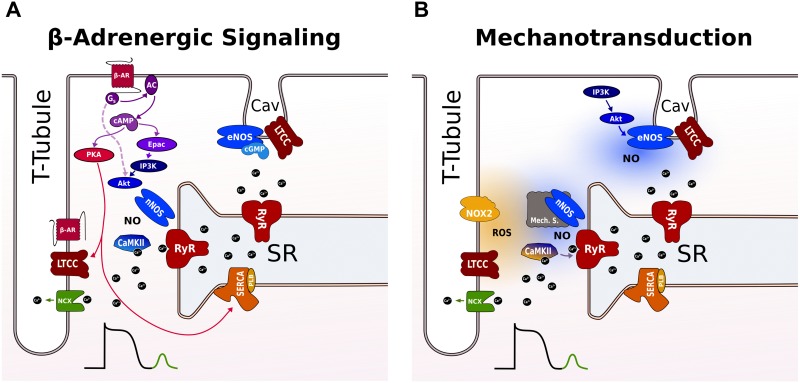
Heart failure (HF) is associated with elevated sympathetic tone and mechanical load. Both systems activate signaling transduction pathways that increase cardiac output, but eventually become part of the disease process itself leading to further worsening of cardiac function. These alterations can adversely contribute to electrical instability, at least in part due to the modulation of Ca2+ handling at the level of the single cardiac myocyte. The major aim of this review is to provide a definitive overview of the links and cross talk between β-adrenergic stimulation, mechanical load, and arrhythmogenesis in the setting of HF. We will initially review the role of Ca2+ in the induction of both early and delayed afterdepolarizations, the role that β-adrenergic stimulation plays in the initiation of these and how the propensity for these may be altered in HF. We will then go onto reviewing the current data with regards to the link between mechanical load and afterdepolarizations, the associated mechano-sensitivity of the ryanodine receptor and other stretch activated channels that may be associated with HF-associated arrhythmias. Furthermore, we will discuss how alterations in local Ca2+ microdomains during the remodeling process associated the HF may contribute to the increased disposition for β-adrenergic or stretch induced arrhythmogenic triggers. Finally, the potential mechanisms linking β-adrenergic stimulation and mechanical stretch will be clarified, with the aim of finding common modalities of arrhythmogenesis that could be targeted by novel therapeutic agents in the setting of HF.
Keywords: calcium; heart failure; microdomains; myocytes; ryanodine; stretch; sympathetic stimulation.
Figures


References
-
- Antoons G., Johnson D. M., Dries E., Santiago D. J., Ozdemir S., Lenaerts I., et al. (2015). Calcium release near l-type calcium channels promotes beat-to-beat variability in ventricular myocytes from the chronic AV block dog. J. Mol. Cell. Cardiol. 89(Part B), 326–334. 10.1016/j.yjmcc.2015.10.008 - DOI - PubMed
-
- Antoons G., Volders P. G., Stankovicova T., Bito V., Stengl M., Vos M. A., et al. (2007). Window Ca2+ current and its modulation by Ca2+ release in hypertrophied cardiac myocytes from dogs with chronic atrioventricular block. J. Physiol. 579 147–160. 10.1113/jphysiol.2006.124222 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous