

Reversal of homocysteine-induced neurotoxicity in rat hippocampal neurons by astaxanthin: evidences for mitochondrial dysfunction and signaling crosstalk
- PMID: 30374413
- PMCID: PMC6197197
- DOI: 10.1038/s41420-018-0114-x
Reversal of homocysteine-induced neurotoxicity in rat hippocampal neurons by astaxanthin: evidences for mitochondrial dysfunction and signaling crosstalk
Erratum in
-
Correction to: Reversal of homocysteine-induced neurotoxicity in rat hippocampal neurons by astaxanthin: evidences for mitochondrial dysfunction and signaling crosstalk.Cell Death Discov. 2019 Mar 1;5:70. doi: 10.1038/s41420-019-0140-3. eCollection 2019. Cell Death Discov. 2019. PMID: 30854229 Free PMC article.
-
Erratum: Publisher Correction: articles initially published in wrong volume.Cell Death Discov. 2019 Jul 10;5:116. doi: 10.1038/s41420-019-0186-2. eCollection 2019. Cell Death Discov. 2019. PMID: 31312525 Free PMC article.
Abstract
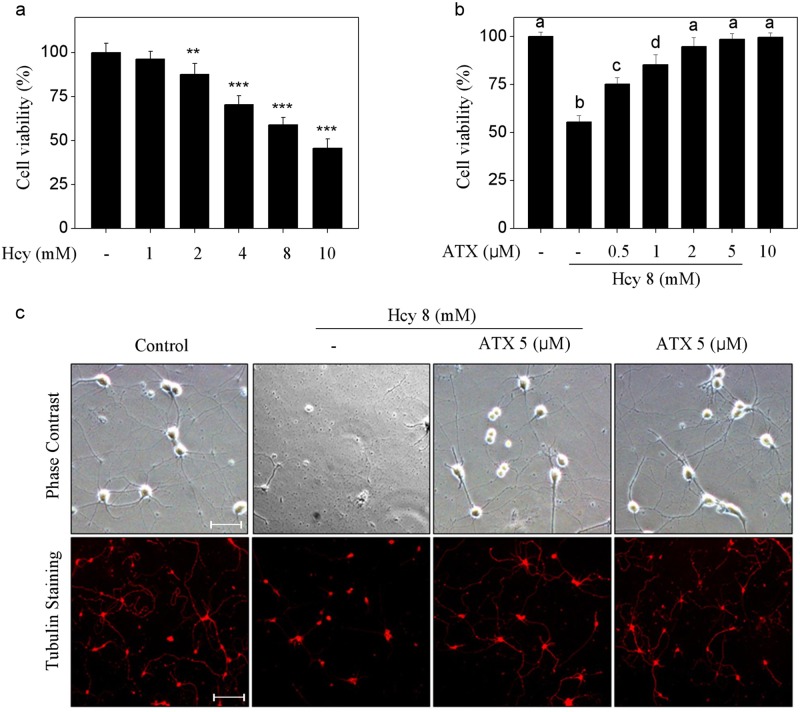
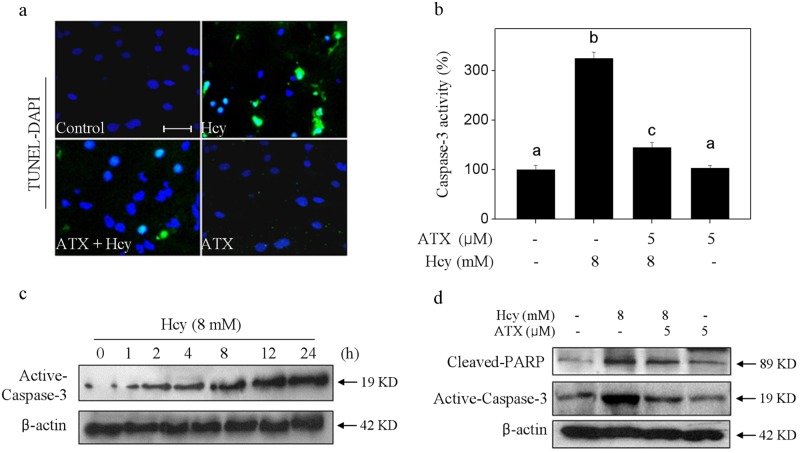
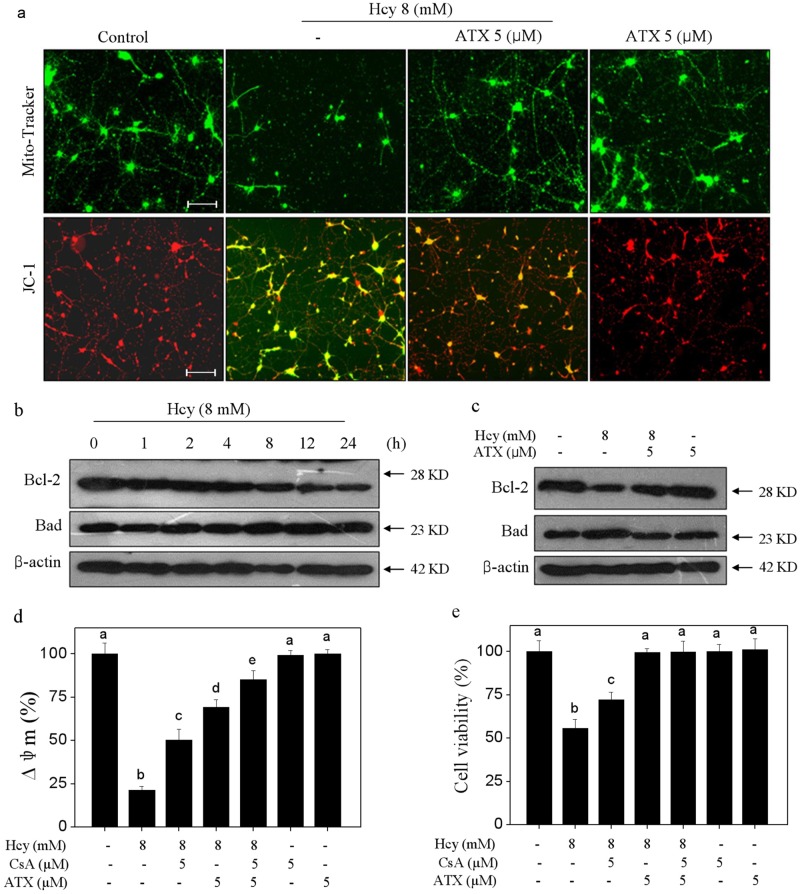
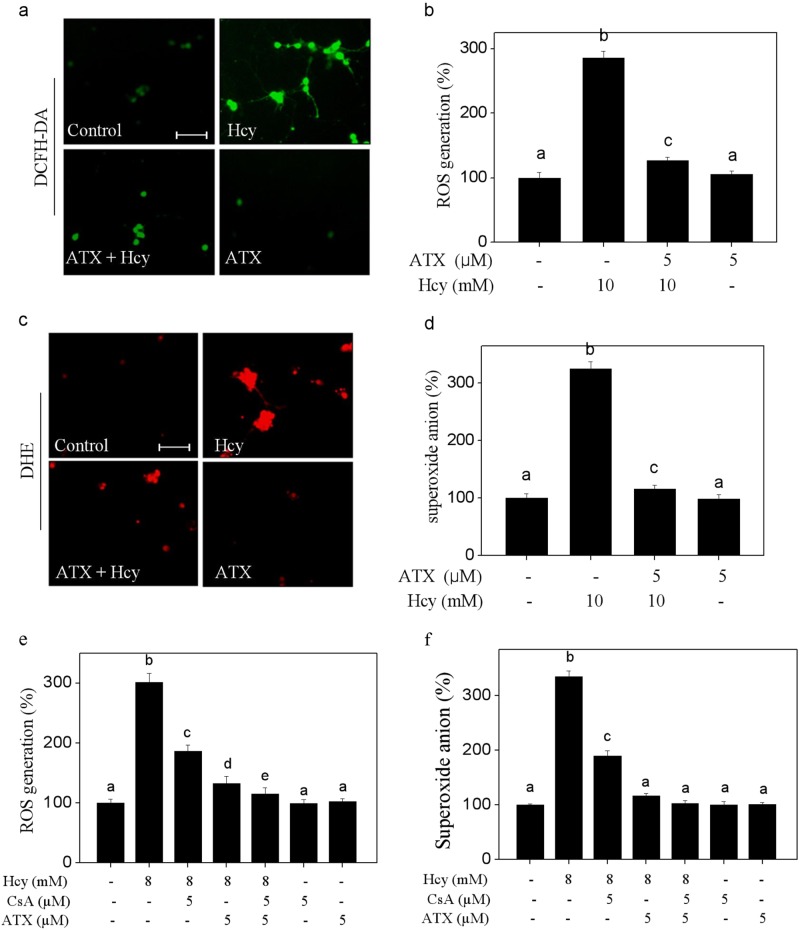
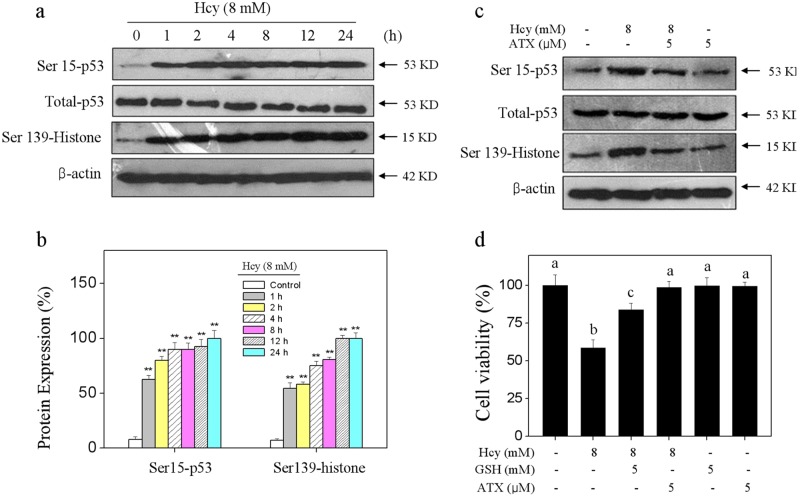
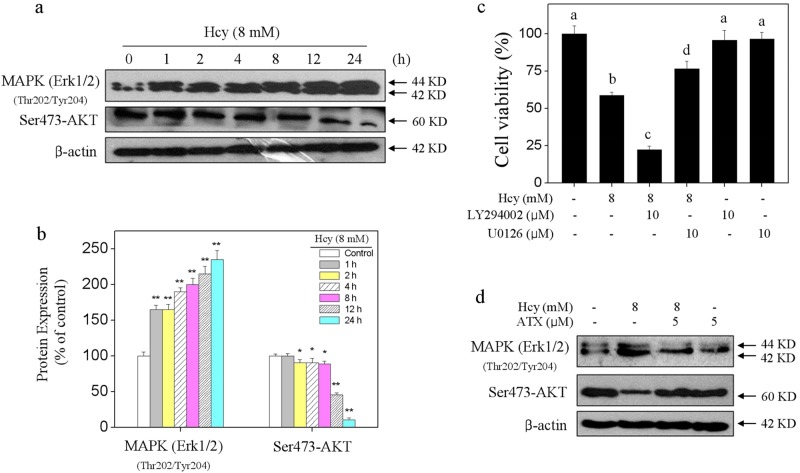
Elevated plasma level of homocysteine (Hcy) represents an independent risk for neurological diseases, and induction of oxidative damage is considered as one of the most important pathomechanisms. Astaxanthin (ATX) exhibits strong antioxidant activity in kinds of experimental models. However, the potential of ATX against Hcy-induced neurotoxicity has not been well explored yet. Herein, the neuroprotective effect of ATX against Hcy-induced neurotoxicity in rat hippocampal neurons was examined, and the underlying mechanism was evaluated. The results showed that ATX pre-treatment completely reversed Hcy-induced neurotoxicity through inhibiting cell apoptosis in rat primary hippocampal neurons. The mechanical investigation revealed that ATX effectively blocked Hcy-induced mitochondrial dysfunction by regulating Bcl-2 family and opening of mitochondrial permeability transition pore (MPTP). ATX pre-treatment also attenuated Hcy-induced oxidative damage via inhibiting the release of intracellular reactive oxide species (ROS) and superoxide anion through regulating MPTP opening. Moreover, normalization of MAPKs and PI3K/AKT pathways also contributed to ATX-mediated protective effects. Taken together, these results above suggested that ATX has the potential to reverse Hcy-induced neurotoxicity and apoptosis by inhibiting mitochondrial dysfunction, ROS-mediated oxidative damage and regulation of MAKPs and AKT pathways, which validated the strategy of using ATX could be a highly effective way in combating Hcy-mediated neurological disorders.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Astaxanthin Attenuates Homocysteine-Induced Cardiotoxicity in Vitro and in Vivo by Inhibiting Mitochondrial Dysfunction and Oxidative Damage.Front Physiol. 2017 Dec 12;8:1041. doi: 10.3389/fphys.2017.01041. eCollection 2017. Front Physiol. 2017. PMID: 29311972 Free PMC article.
-
Hydrogen sulfide attenuates homocysteine-induced neurotoxicity by preventing mitochondrial dysfunctions and oxidative damage: In vitro and in vivo studies.Neurochem Int. 2018 Nov;120:87-98. doi: 10.1016/j.neuint.2018.07.010. Epub 2018 Jul 26. Neurochem Int. 2018. PMID: 30055195
-
Neuroprotective effect of astaxanthin against glutamate-induced cytotoxicity in HT22 cells: Involvement of the Akt/GSK-3β pathway.Neuroscience. 2015 Sep 10;303:558-68. doi: 10.1016/j.neuroscience.2015.07.034. Epub 2015 Jul 18. Neuroscience. 2015. PMID: 26197224
-
Astaxanthin Protects PC12 Cells against Homocysteine- and Glutamate-Induced Neurotoxicity.Molecules. 2020 Jan 5;25(1):214. doi: 10.3390/molecules25010214. Molecules. 2020. PMID: 31948056 Free PMC article.
-
Astaxanthin upregulates heme oxygenase-1 expression through ERK1/2 pathway and its protective effect against beta-amyloid-induced cytotoxicity in SH-SY5Y cells.Brain Res. 2010 Nov 11;1360:159-67. doi: 10.1016/j.brainres.2010.08.100. Epub 2010 Sep 7. Brain Res. 2010. PMID: 20828541
Cited by
-
The Homocysteine and Metabolic Syndrome: A Mendelian Randomization Study.Nutrients. 2021 Jul 16;13(7):2440. doi: 10.3390/nu13072440. Nutrients. 2021. PMID: 34371949 Free PMC article.
-
Formulation Approaches to Crystalline Status Modification for Carotenoids: Impacts on Dissolution, Stability, Bioavailability, and Bioactivities.Pharmaceutics. 2023 Feb 1;15(2):485. doi: 10.3390/pharmaceutics15020485. Pharmaceutics. 2023. PMID: 36839810 Free PMC article. Review.
-
Astaxanthin Modulation of Signaling Pathways That Regulate Autophagy.Mar Drugs. 2019 Sep 23;17(10):546. doi: 10.3390/md17100546. Mar Drugs. 2019. PMID: 31547619 Free PMC article. Review.
-
The Role of Dietary Antioxidants in the Pathogenesis of Neurodegenerative Diseases and Their Impact on Cerebral Oxidoreductive Balance.Nutrients. 2020 Feb 8;12(2):435. doi: 10.3390/nu12020435. Nutrients. 2020. PMID: 32046360 Free PMC article. Review.
-
Hexavalent Chromium Induces Neurotoxicity by Triggering Mitochondrial Dysfunction and ROS-Mediated Signals.Neurochem Res. 2024 Mar;49(3):660-669. doi: 10.1007/s11064-023-04063-y. Epub 2023 Nov 27. Neurochem Res. 2024. PMID: 38010603
References
-
- Rozycka A, Jagodzinski PP, Kozubski W, Lianeri M, Dorszewska J. Homocysteine level and mechanisms of injury in Parkinson’s disease as related to MTHFR, MTR, and MTHFD1 genes polymorphisms and L-Dopa treatment. Curr. Genom. 2013;14:534–542. doi: 10.2174/1389202914666131210210559. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
