

Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure
- PMID: 30375404
- PMCID: PMC6207673
- DOI: 10.1038/s41467-018-06639-7
Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure
Abstract
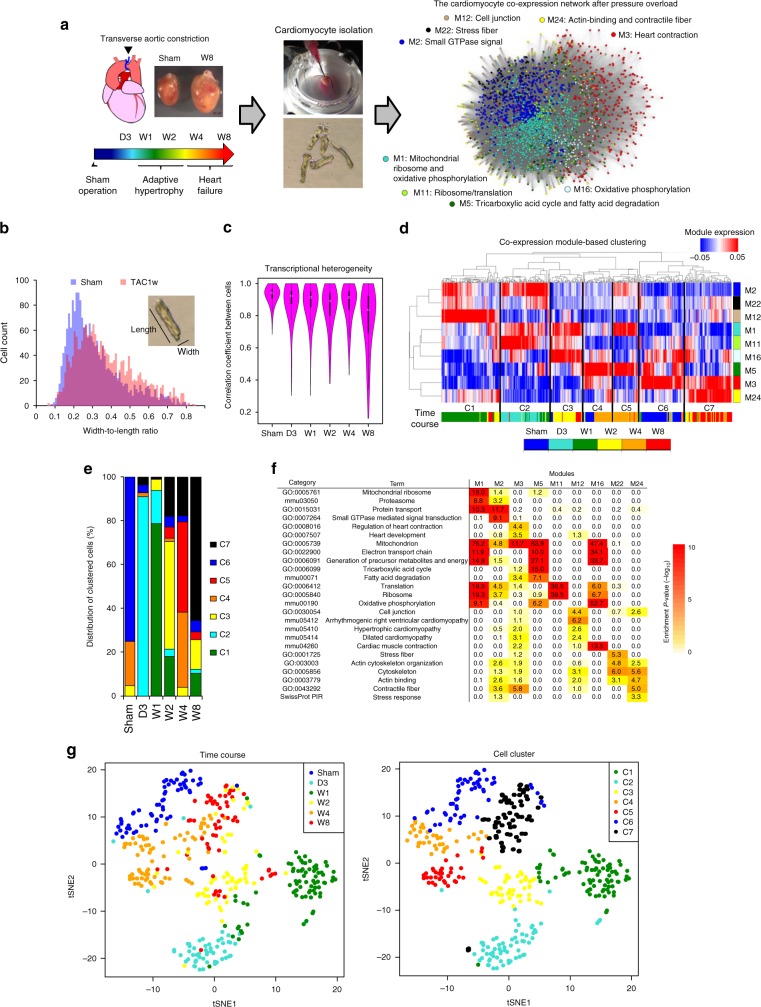
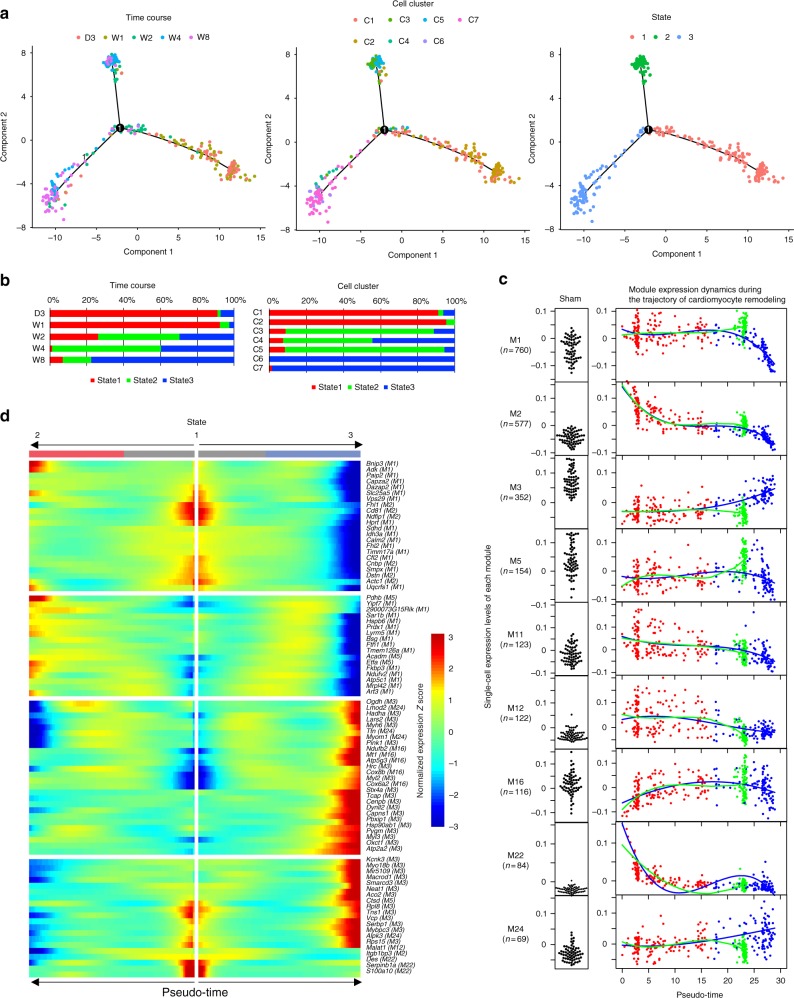
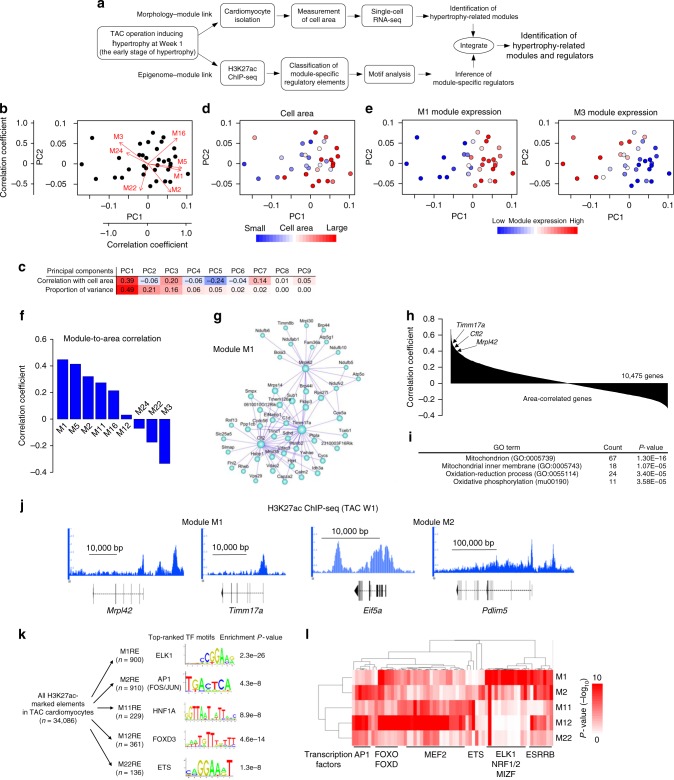
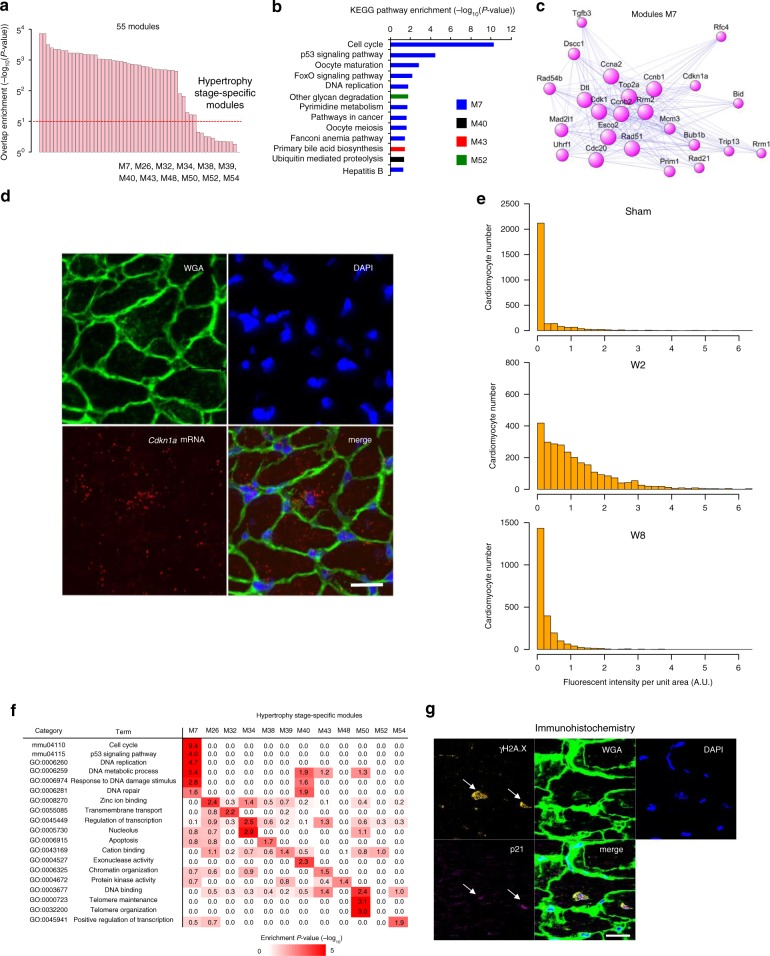
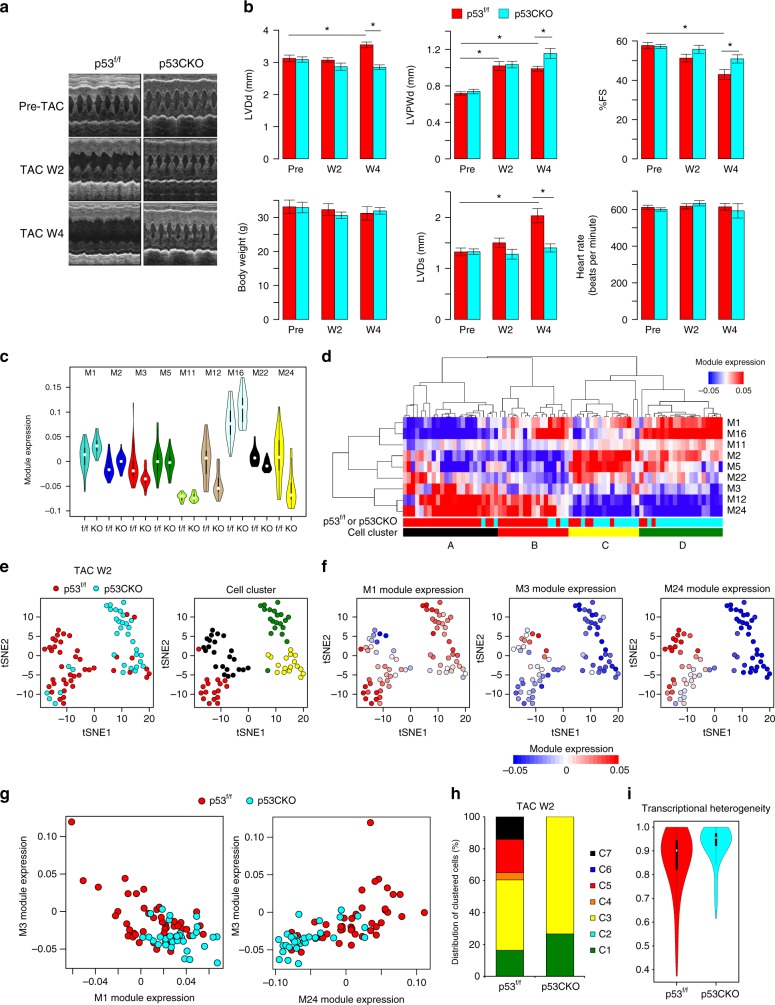
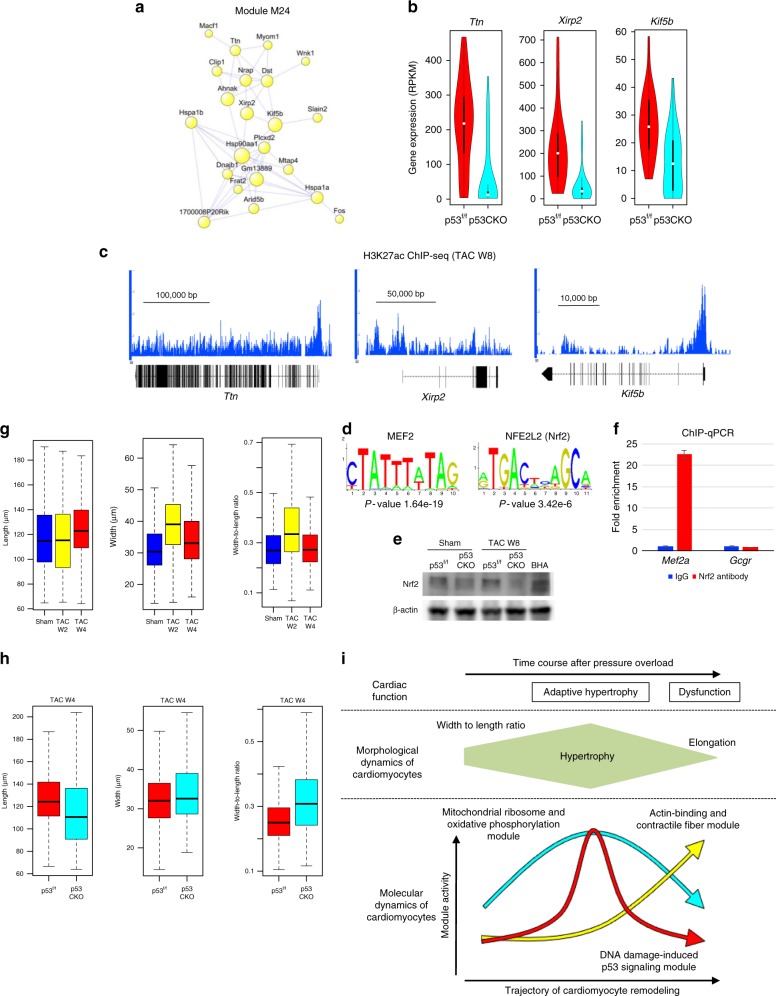
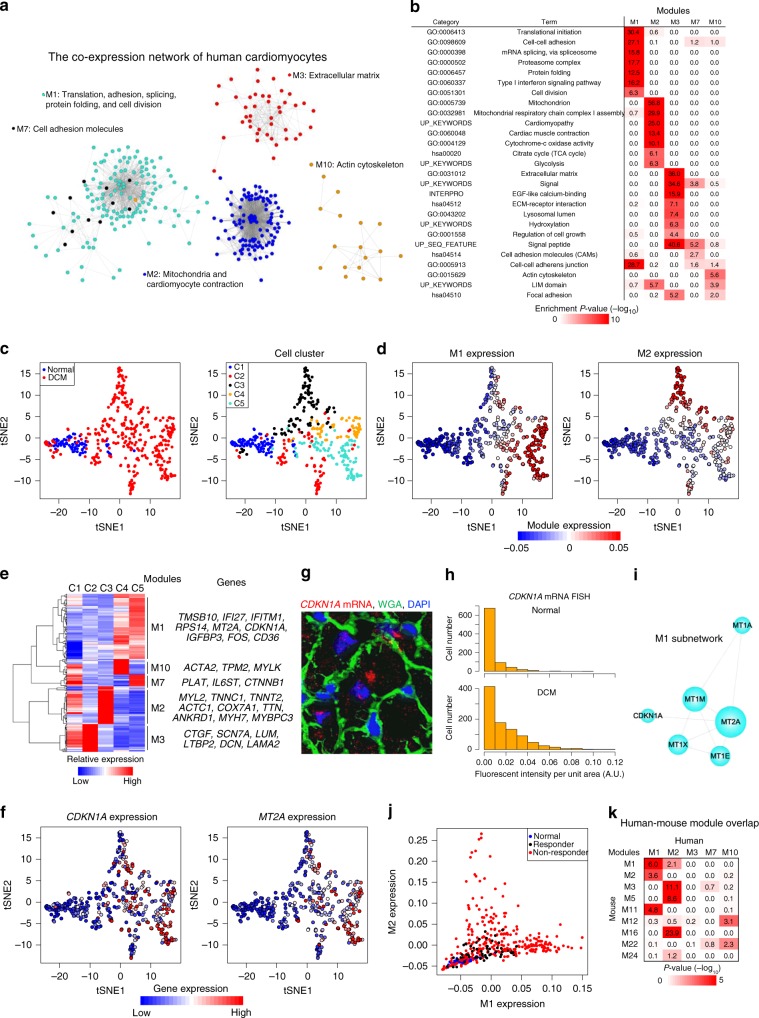
Pressure overload induces a transition from cardiac hypertrophy to heart failure, but its underlying mechanisms remain elusive. Here we reconstruct a trajectory of cardiomyocyte remodeling and clarify distinct cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure, by integrating single-cardiomyocyte transcriptome with cell morphology, epigenomic state and heart function. During early hypertrophy, cardiomyocytes activate mitochondrial translation/metabolism genes, whose expression is correlated with cell size and linked to ERK1/2 and NRF1/2 transcriptional networks. Persistent overload leads to a bifurcation into adaptive and failing cardiomyocytes, and p53 signaling is specifically activated in late hypertrophy. Cardiomyocyte-specific p53 deletion shows that cardiomyocyte remodeling is initiated by p53-independent mitochondrial activation and morphological hypertrophy, followed by p53-dependent mitochondrial inhibition, morphological elongation, and heart failure gene program activation. Human single-cardiomyocyte analysis validates the conservation of the pathogenic transcriptional signatures. Collectively, cardiomyocyte identity is encoded in transcriptional programs that orchestrate morphological and functional phenotypes.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
The alteration of protein prenylation induces cardiomyocyte hypertrophy through Rheb-mTORC1 signalling and leads to chronic heart failure.J Pathol. 2015 Apr;235(5):672-85. doi: 10.1002/path.4480. Epub 2015 Jan 7. J Pathol. 2015. PMID: 25385233
-
The H19 long noncoding RNA is a novel negative regulator of cardiomyocyte hypertrophy.Cardiovasc Res. 2016 Jul 1;111(1):56-65. doi: 10.1093/cvr/cvw078. Epub 2016 Apr 15. Cardiovasc Res. 2016. PMID: 27084844
-
p53 regulates the cardiac transcriptome.Proc Natl Acad Sci U S A. 2017 Feb 28;114(9):2331-2336. doi: 10.1073/pnas.1621436114. Epub 2017 Feb 13. Proc Natl Acad Sci U S A. 2017. PMID: 28193895 Free PMC article.
-
How cardiomyocytes sense pathophysiological stresses for cardiac remodeling.Cell Mol Life Sci. 2017 Mar;74(6):983-1000. doi: 10.1007/s00018-016-2373-0. Epub 2016 Oct 6. Cell Mol Life Sci. 2017. PMID: 27714411 Free PMC article. Review.
-
Gene expression profiles, potential targets and treatments of cardiac remodeling.Mol Cell Biochem. 2025 Mar;480(3):1555-1567. doi: 10.1007/s11010-024-05126-6. Epub 2024 Oct 5. Mol Cell Biochem. 2025. PMID: 39367915 Review.
Cited by
-
Single-cell and spatial transcriptomics approaches of cardiovascular development and disease.BMB Rep. 2020 Aug;53(8):393-399. doi: 10.5483/BMBRep.2020.53.8.130. BMB Rep. 2020. PMID: 32684243 Free PMC article. Review.
-
Role of miRNA-1 and miRNA-21 in Acute Myocardial Ischemia-Reperfusion Injury and Their Potential as Therapeutic Strategy.Int J Mol Sci. 2022 Jan 28;23(3):1512. doi: 10.3390/ijms23031512. Int J Mol Sci. 2022. PMID: 35163436 Free PMC article. Review.
-
Applications of Single-Cell RNA Sequencing in Cardiovascular Research.Front Cell Dev Biol. 2022 Jan 31;9:810232. doi: 10.3389/fcell.2021.810232. eCollection 2021. Front Cell Dev Biol. 2022. PMID: 35174168 Free PMC article. Review.
-
TEAD1 trapping by the Q353R-Lamin A/C causes dilated cardiomyopathy.Sci Adv. 2023 Apr 14;9(15):eade7047. doi: 10.1126/sciadv.ade7047. Epub 2023 Apr 14. Sci Adv. 2023. PMID: 37058558 Free PMC article.
-
Genetic and phenotypic architecture of human myocardial trabeculation.Nat Cardiovasc Res. 2024 Dec;3(12):1503-1515. doi: 10.1038/s44161-024-00564-3. Epub 2024 Nov 20. Nat Cardiovasc Res. 2024. PMID: 39567769 Free PMC article.
References
-
- Mann DL, Felker GM. Heart Failure: A Companion to Braunwald’s Heart Disease. 3rd edn. Amsterdam: Elsevier; 2016.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
