

Histological heterogeneity in a large clinical cohort of juvenile idiopathic inflammatory myopathy: analysis by myositis autoantibody and pathological features
- PMID: 30378704
- PMCID: PMC6767402
- DOI: 10.1111/nan.12528
Histological heterogeneity in a large clinical cohort of juvenile idiopathic inflammatory myopathy: analysis by myositis autoantibody and pathological features
Abstract
Aim: Juvenile idiopathic inflammatory myopathies have been recently reclassified into clinico-serological subgroups. Myopathological correlates of the subgroups are incompletely understood.
Methods: We studied muscle biopsies from 101 children with clinically and serologically defined juvenile idiopathic inflammatory myopathies from the UK JDM Cohort and Biomarker Study by applying the international JDM score tool, myopathological review and C5b-9 complement analysis.
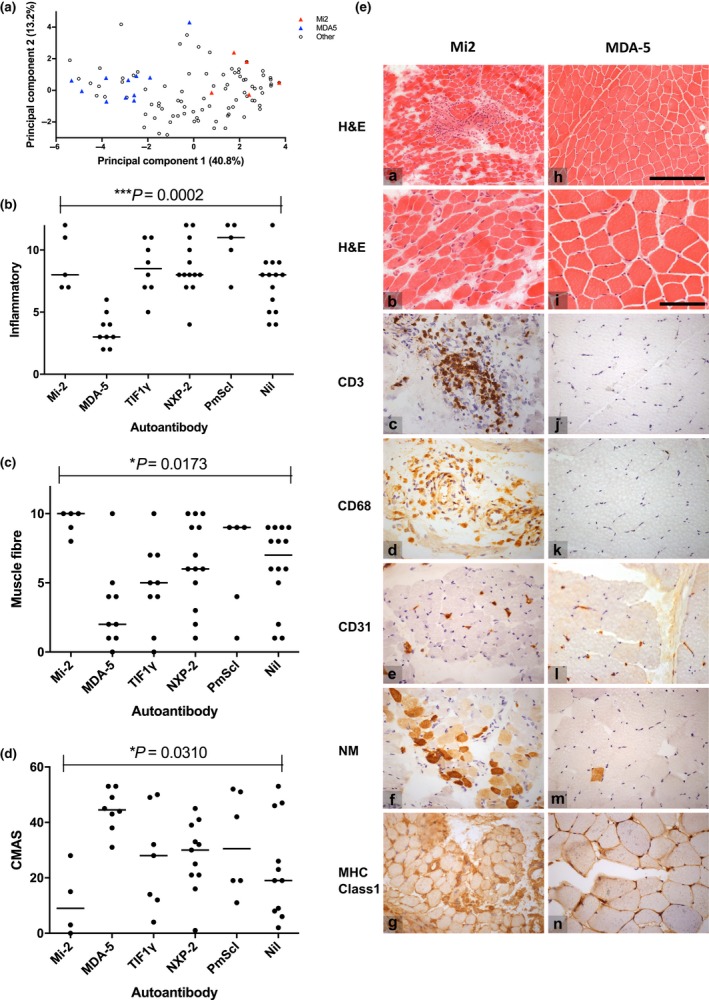
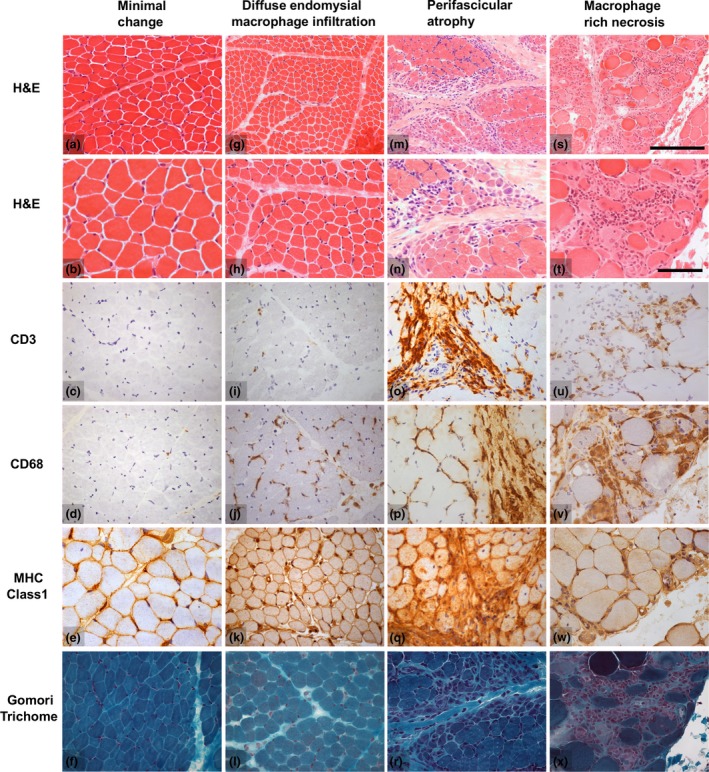
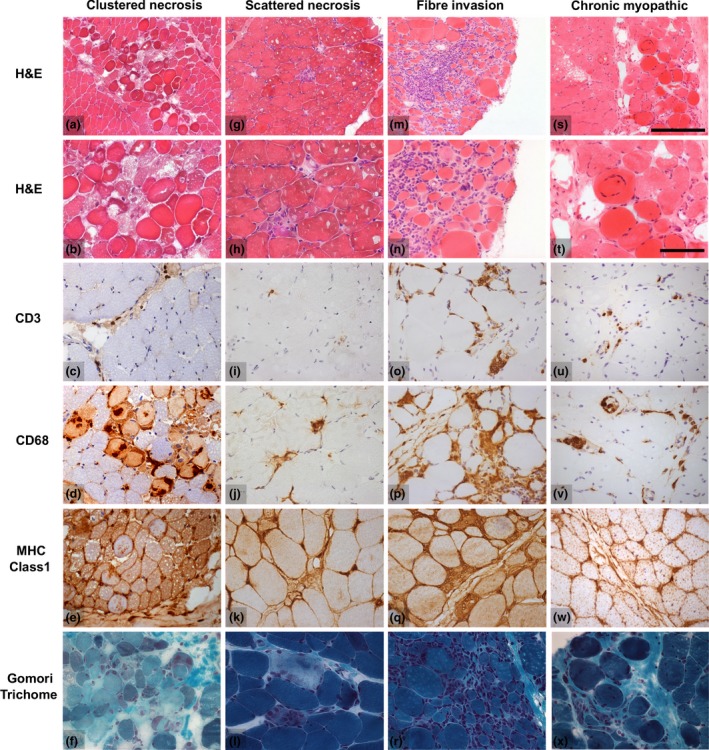
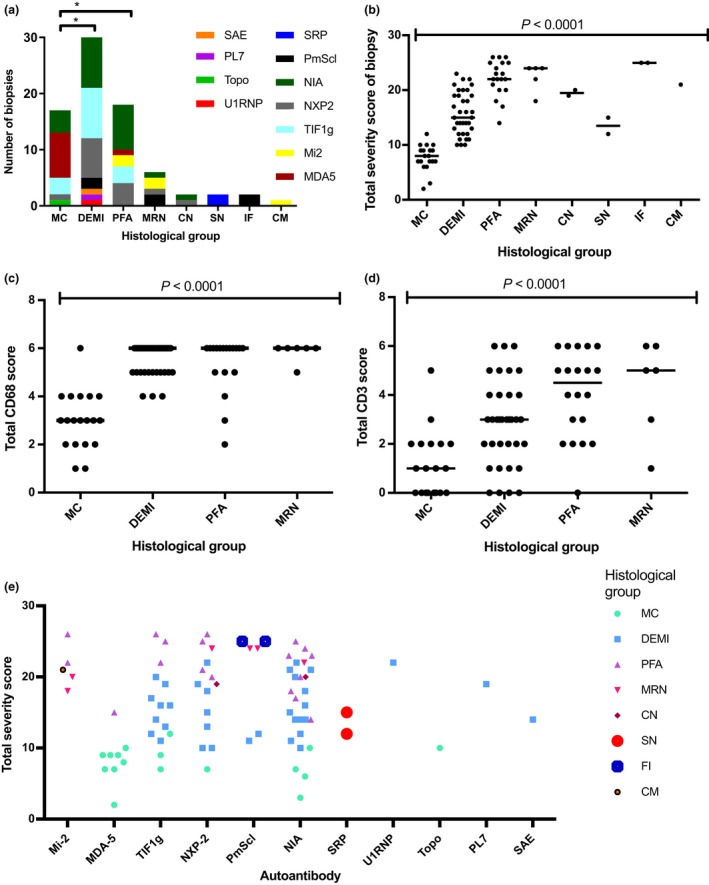
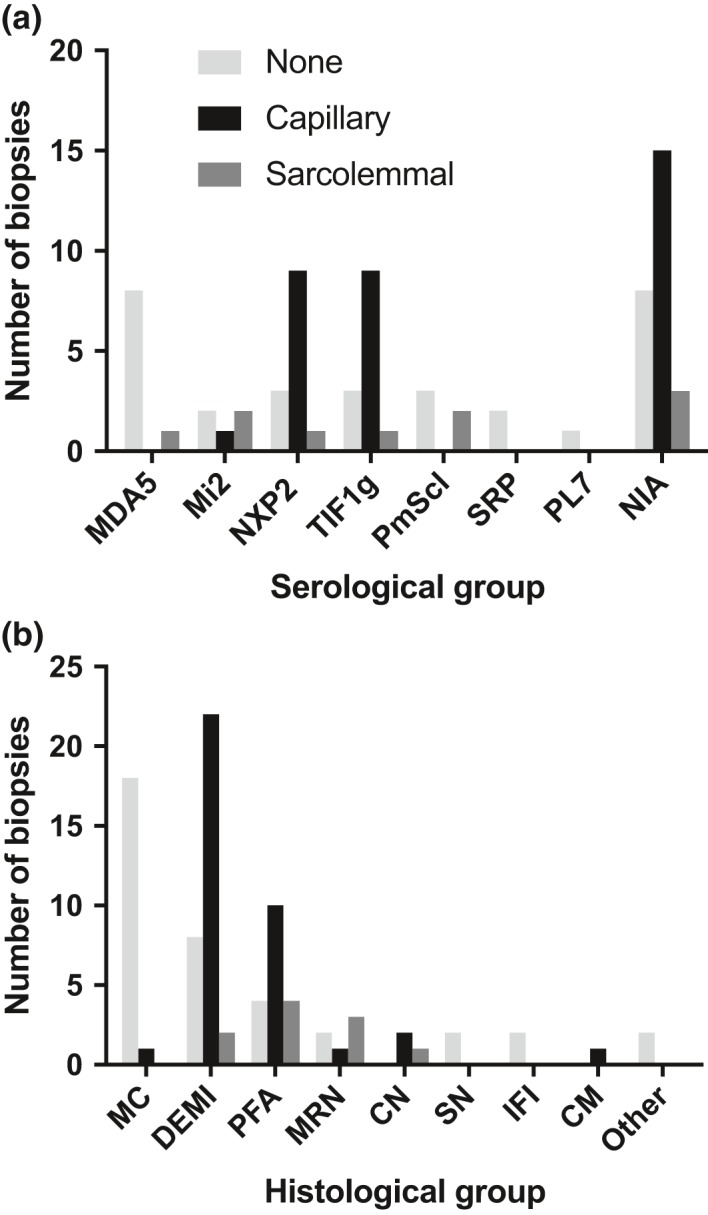
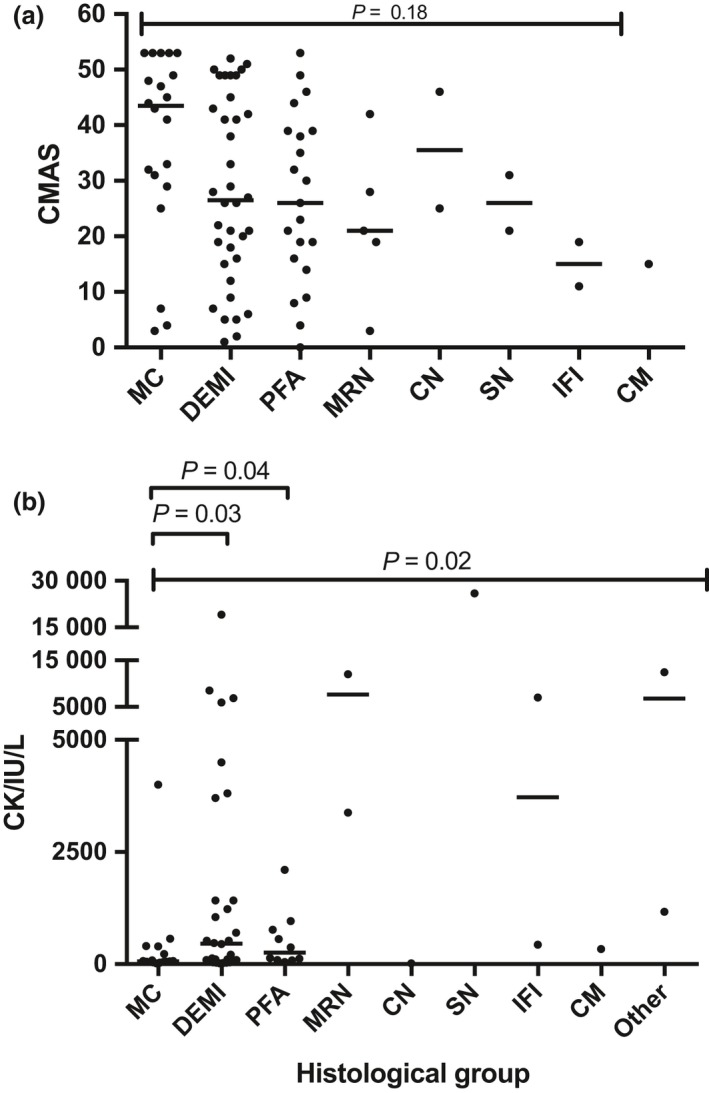
Results: Autoantibody data were available for 90/101 cases with 18/90 cases positive for anti-TIF1γ, 15/90 anti-NXP2, 11/90 anti-MDA5, 5/90 anti-Mi2 and 6/90 anti-PmScl. JDM biopsy severity scores were consistently low in the anti-MDA5 group, high in the anti-Mi2 group, and widely distributed in the other groups. Biopsies were classified histologically as perifascicular atrophy (22/101), macrophage-rich necrosis (6/101), scattered necrosis (2/101), clustered necrosis (2/101), inflammatory fibre invasion (2/101), chronic myopathic change (1/101), diffuse endomysial macrophage infiltrates (40/101) and minimal change (24/101). MDA5 cases segregated with the minimal change group and showed no capillary C5b-9-deposition. The Mi2 group displayed high severity scores and a tendency towards sarcolemmal complement deposition. NXP2 and TIF1γ groups showed a variety of pathologies with a high proportion of diffuse endomysial macrophage infiltrates and a high proportion of capillary C5b-9 deposition.
Conclusion: We have shown that juvenile idiopathic inflammatory myopathies have a spectrum of histopathological phenotypes and show distinct complement attack complex deposition patterns. Both correlate in some cases with the serological subtypes. Most cases do not show typical histological features associated with dermatomyositis (e.g. perifascicular atrophy). In contrast, more than half show relatively mild histopathological changes.
Keywords: JDM score tool; anti-SRP; dermatomyositis; immune mediated necrotizing myopathy; polymyositis; principal component analysis.
© 2019 The Authors. Neuropathology and Applied Neurobiology published by John Wiley & Sons Ltd on behalf of British Neuropathological Society.
Conflict of interest statement
The authors declare no conflict of interest. The Editors of
Figures
References
-
- Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, et al 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004; 14: 337–45 - PubMed
-
- Mescam‐Mancini L, Allenbach Y, Hervier B, Devilliers H, Mariampillay K, Dubourg O, et al Anti‐Jo‐1 antibody‐positive patients show a characteristic necrotizing perifascicular myositis. Brain 2015; 138: 2485–92 - PubMed
-
- Stenzel W, Preusse C, Allenbach Y, Pehl D, Junckerstorff R, Heppner FL, et al Nuclear actin aggregation is a hallmark of anti‐synthetase syndrome‐induced dysimmune myopathy. Neurology 2015; 84: 1346–54 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
