

Whole-Genome Alignment and Comparative Annotation
- PMID: 30379572
- PMCID: PMC6450745
- DOI: 10.1146/annurev-animal-020518-115005
Whole-Genome Alignment and Comparative Annotation
Abstract
Rapidly improving sequencing technology coupled with computational developments in sequence assembly are making reference-quality genome assembly economical. Hundreds of vertebrate genome assemblies are now publicly available, and projects are being proposed to sequence thousands of additional species in the next few years. Such dense sampling of the tree of life should give an unprecedented new understanding of evolution and allow a detailed determination of the events that led to the wealth of biodiversity around us. To gain this knowledge, these new genomes must be compared through genome alignment (at the sequence level) and comparative annotation (at the gene level). However, different alignment and annotation methods have different characteristics; before starting a comparative genomics analysis, it is important to understand the nature of, and biases and limitations inherent in, the chosen methods. This review is intended to act as a technical but high-level overview of the field that should provide this understanding. We briefly survey the state of the genome alignment and comparative annotation fields and potential future directions for these fields in a new, large-scale era of comparative genomics.
Keywords: comparative genomics; genome alignment; genome annotation.
Figures
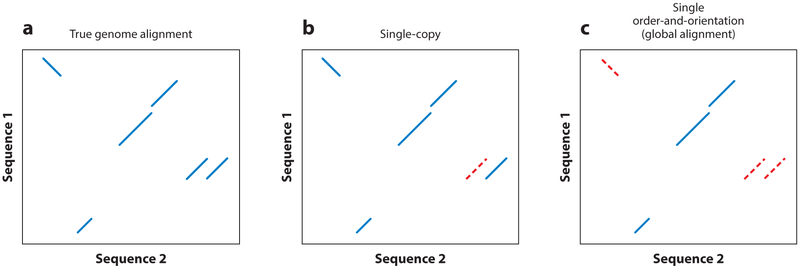
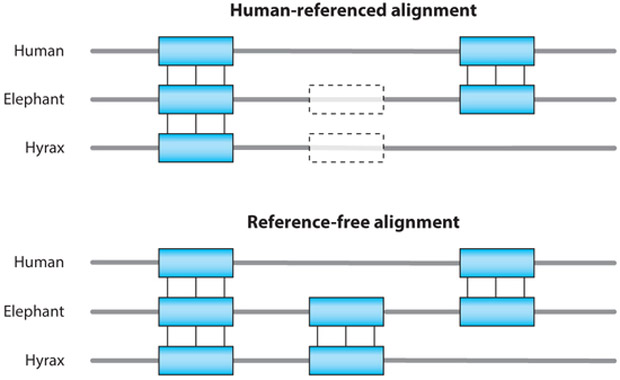
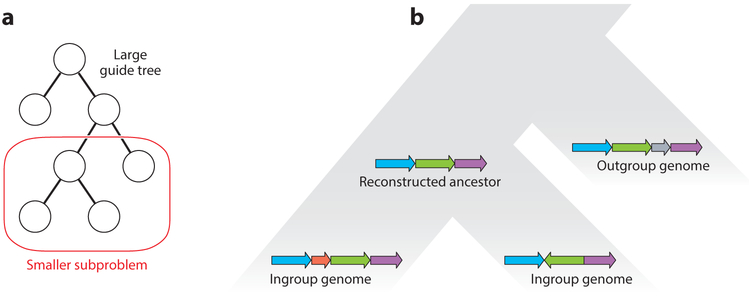
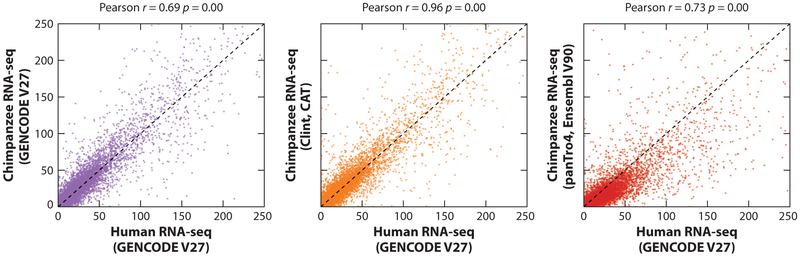
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources