

Steered molecular dynamics simulations reveal critical residues for (un)binding of substrates, inhibitors and a product to the malarial M1 aminopeptidase
- PMID: 30379805
- PMCID: PMC6239339
- DOI: 10.1371/journal.pcbi.1006525
Steered molecular dynamics simulations reveal critical residues for (un)binding of substrates, inhibitors and a product to the malarial M1 aminopeptidase
Abstract
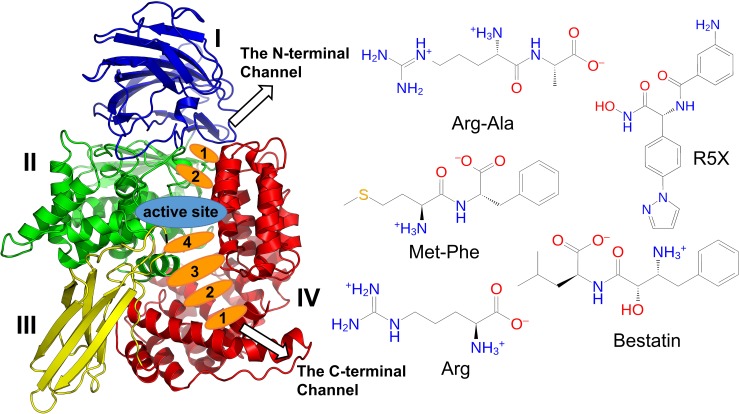
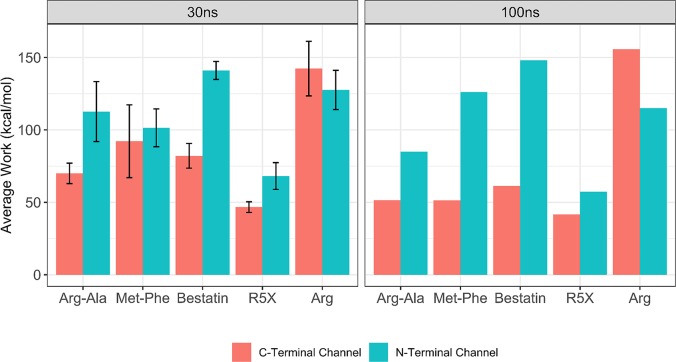
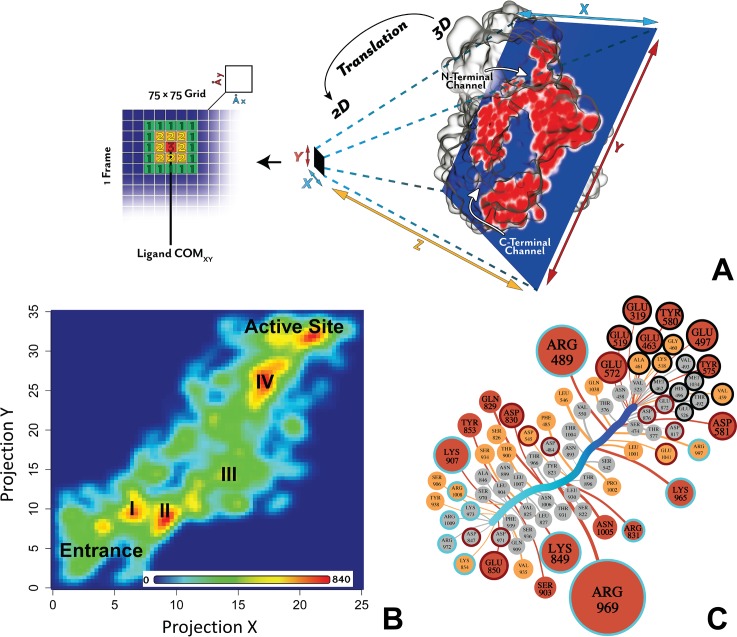
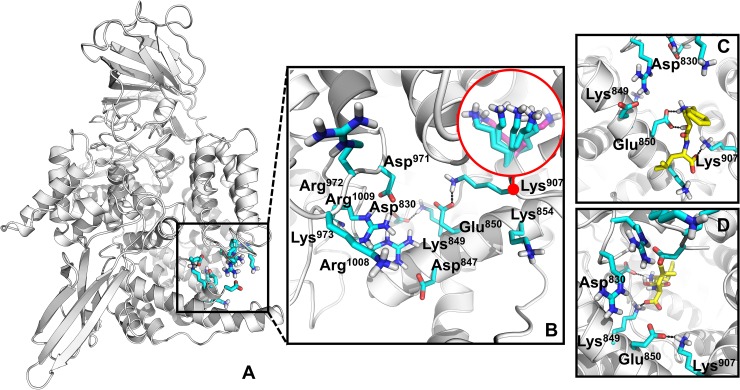
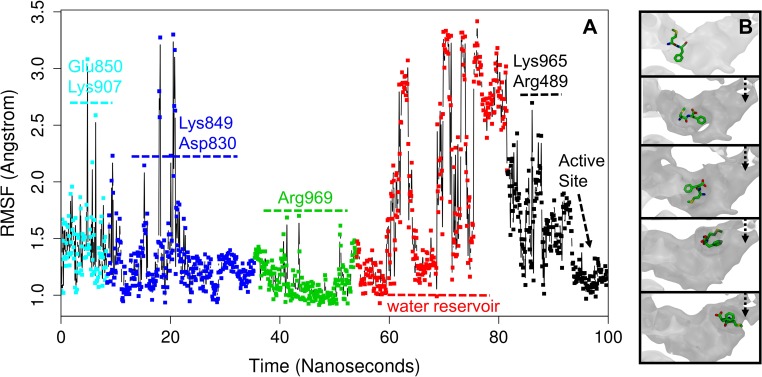
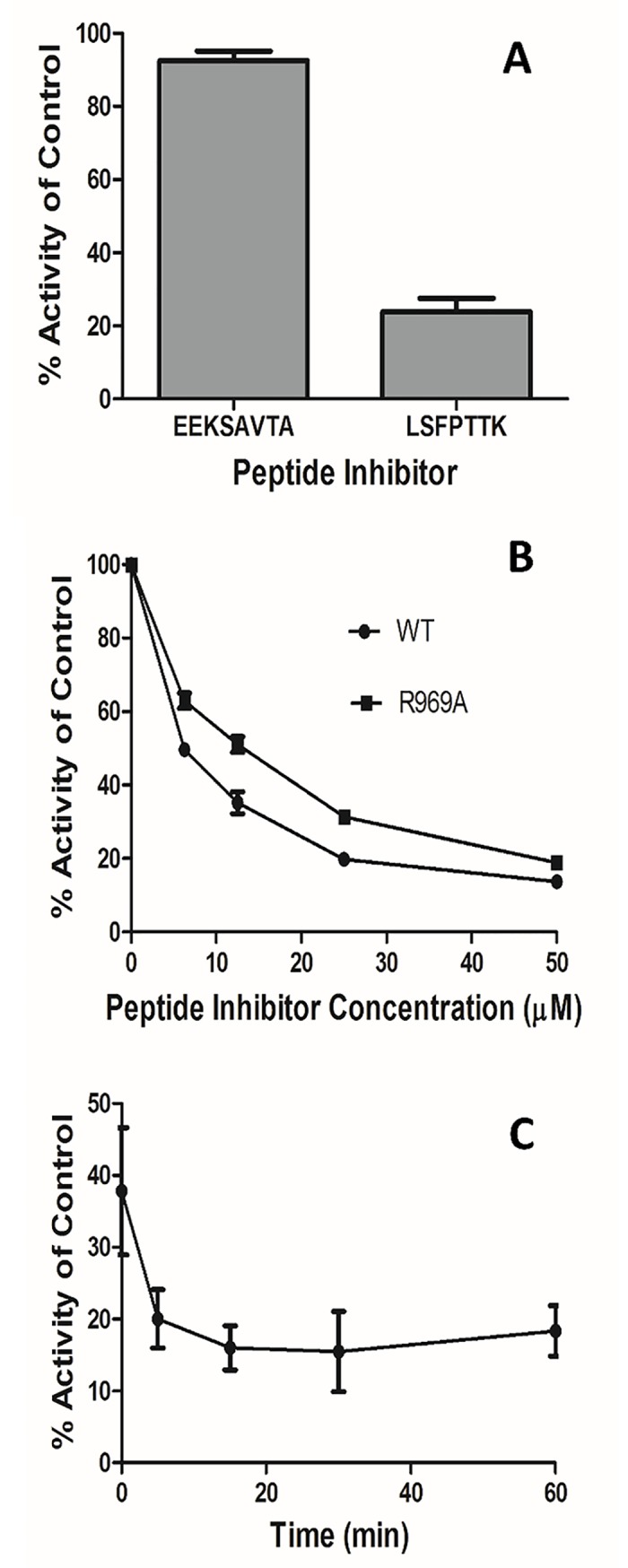
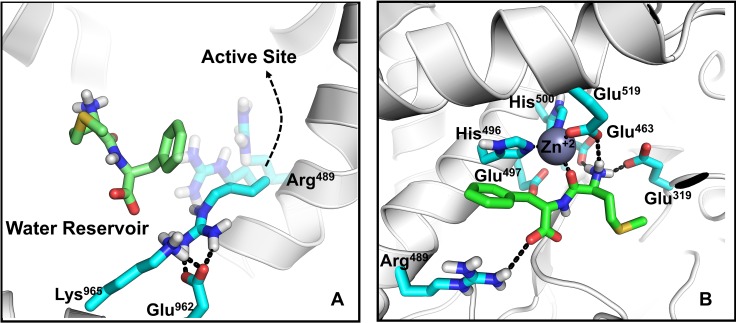
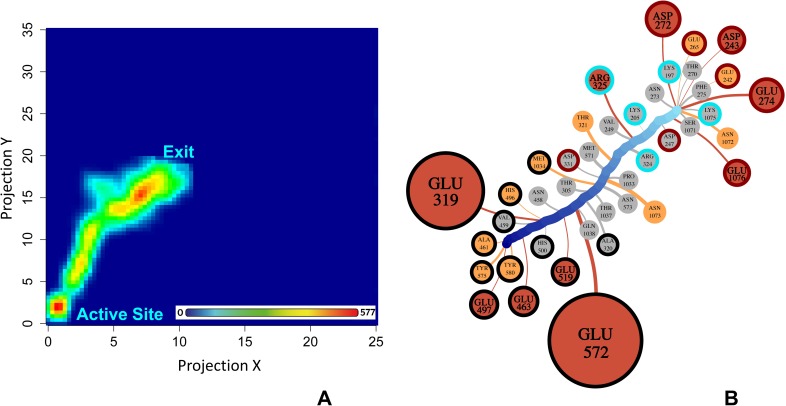
Malaria is a life-threatening disease spread by mosquitoes. Plasmodium falciparum M1 alanyl aminopeptidase (PfM1-AAP) is a promising target for the treatment of malaria. The recently solved crystal structures of PfM1-AAP revealed that the buried active site can be accessed through two channel openings: a short N-terminal channel with the length of 8 Å and a long C-terminal channel with the length of 30 Å. It is unclear, however, how substrates and inhibitors migrate to the active site and a product of cleavage leaves. Here, we study the molecular mechanism of substrate and inhibitor migration to the active site and the product release using steered molecular dynamics simulations. We identified a stepwise passage of substrates and inhibitors in the C-terminal channel of PfM1-AAP, involving (I) ligand recognition at the opening of the channel, (II) ionic translation to the 'water reservoir', (III) ligand reorientation in the 'water reservoir' and (IV) passage in a suitable conformation into the active site. Endorsed by enzymatic analysis of functional recombinant PfM1-AAP and mutagenesis studies, our novel ligand-residue binding network analysis has identified the functional residues controlling ligand migration within the C-terminal channel of PfM1-AAP. Furthermore, from unbinding simulations of the Arg product we propose a charge repulsion as the driving force to expel the product out from the N-terminal channel of PfM1-AAP. Our work paves the way towards the design of a novel class of PfM1-AAP inhibitors based on preventing substrate entry to the active site.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



Similar articles
-
X-ray crystal structure and specificity of the Plasmodium falciparum malaria aminopeptidase PfM18AAP.J Mol Biol. 2012 Sep 28;422(4):495-507. doi: 10.1016/j.jmb.2012.06.006. Epub 2012 Jun 16. J Mol Biol. 2012. PMID: 22709581
-
The Plasmodium falciparum malaria M1 alanyl aminopeptidase (PfA-M1): insights of catalytic mechanism and function from MD simulations.PLoS One. 2011;6(12):e28589. doi: 10.1371/journal.pone.0028589. Epub 2011 Dec 21. PLoS One. 2011. PMID: 22205955 Free PMC article.
-
Structure of the Plasmodium falciparum M17 aminopeptidase and significance for the design of drugs targeting the neutral exopeptidases.Proc Natl Acad Sci U S A. 2010 Feb 9;107(6):2449-54. doi: 10.1073/pnas.0911813107. Epub 2010 Jan 21. Proc Natl Acad Sci U S A. 2010. PMID: 20133789 Free PMC article.
-
Mapping the Pathway and Dynamics of Bestatin Inhibition of the Plasmodium falciparum M1 Aminopeptidase PfA-M1.ChemMedChem. 2018 Dec 6;13(23):2504-2513. doi: 10.1002/cmdc.201800563. Epub 2018 Nov 9. ChemMedChem. 2018. PMID: 30318749
-
Plasmodium falciparum neutral aminopeptidases: new targets for anti-malarials.Trends Biochem Sci. 2010 Jan;35(1):53-61. doi: 10.1016/j.tibs.2009.08.004. Epub 2009 Sep 30. Trends Biochem Sci. 2010. PMID: 19796954 Review.
Cited by
-
Biochemical and cellular characterisation of the Plasmodium falciparum M1 alanyl aminopeptidase (PfM1AAP) and M17 leucyl aminopeptidase (PfM17LAP).Sci Rep. 2021 Feb 3;11(1):2854. doi: 10.1038/s41598-021-82499-4. Sci Rep. 2021. PMID: 33536500 Free PMC article.
-
Chemoproteomics validates selective targeting of Plasmodium M1 alanyl aminopeptidase as an antimalarial strategy.Elife. 2024 Jul 8;13:RP92990. doi: 10.7554/eLife.92990. Elife. 2024. PMID: 38976500 Free PMC article.
-
Random acceleration and steered molecular dynamics simulations reveal the (un)binding tunnels in adenosine deaminase and critical residues in tunnels.RSC Adv. 2020 Dec 11;10(72):43994-44002. doi: 10.1039/d0ra07796h. eCollection 2020 Dec 9. RSC Adv. 2020. PMID: 35517169 Free PMC article.
-
Role of salt-bridging interactions in recognition of viral RNA by arginine-rich peptides.Biophys J. 2021 Nov 16;120(22):5060-5073. doi: 10.1016/j.bpj.2021.10.007. Epub 2021 Oct 26. Biophys J. 2021. PMID: 34710377 Free PMC article.
References
-
- World Malaria Report 2016: Summary. Geneva: World Health Organization; 2017 (WHO/HTM/GMP/2017.4). Licence: CC BY-NC-SA 3.0 IGO.
-
- Mok S, Ashley EA, Ferreira PE, Zhu L, Lin Z, Yeo T, et al. Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 2015. January 23;347(6220):431–435. 10.1126/science.1260403 - DOI - PMC - PubMed
-
- Flannery EL, Chatterjee AK, Winzeler EA. Antimalarial drug discovery—approaches and progress towards new medicines. Nat Rev Microbiol 2013. December;11(12):849–862. 10.1038/nrmicro3138 - DOI - PMC - PubMed
-
- Rosenthal PJ, Sijwali PS, Singh A, Shenai BR. Cysteine proteases of malaria parasites: targets for chemotherapy. Curr Pharm Des 2002;8(18):1659–1672. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
